

Paracetamol
Encyclopedia
Paracetamol INN
(icon or ˌ), or acetaminophen USAN
, is a widely used over-the-counter
analgesic
(pain reliever) and antipyretic
(fever reducer). It is commonly used for the relief of headache
s and other minor aches and pains and is a major ingredient in numerous cold
and flu
remedies. In combination with opioid analgesics, paracetamol can also be used in the management of more severe pain such as post surgical pain and providing palliative care
in advanced cancer patients. The onset of analgesia is approximately 11 minutes after oral administration
of paracetamol, and its half-life
is 1–4 hours.
While generally safe for use at recommended doses ( and up to for adults), acute overdoses of paracetamol can cause potentially fatal liver damage
and, in rare individuals, a normal dose can do the same; the risk is heightened by alcohol consumption
. Paracetamol toxicity
is the foremost cause of acute liver failure in the Western world
, and accounts for most drug overdoses in the United States, the United Kingdom, Australia and New Zealand.
It is the active metabolite of phenacetin
, once popular as an analgesic and antipyretic in its own right, but unlike phenacetin and its combinations, paracetamol is not considered carcinogen
ic at therapeutic doses. The words acetaminophen (used in the United States, Canada, Japan, South Korea, Hong Kong, and Iran) and paracetamol (used elsewhere) both come from a chemical name for the compound: para-acetylaminophenol and para-acetylaminophenol. In some contexts, it is simply abbreviated as APAP, for acetyl-para-aminophenol.
in people of all ages. The World Health Organization
(WHO) recommends that paracetamol only be used to treat fever in children if their temperature is greater than 38.5 °C (101.3 °F). The efficacy of paracetamol by itself in children with fevers has been questioned and a meta-analysis showed that it is less effective than ibuprofen
.
Paracetamol has a well-established role in pediatric medicine as an effective analgesic and antipyretic.
properties comparable to those of aspirin
, while its anti-inflammatory effects are weaker. It is better tolerated than aspirin in patients in whom excessive gastric acid
secretion or prolongation of bleeding time may be a concern. Available without a prescription, it has in recent years increasingly become a common household drug.
Paracetamol can relieve pain in mild arthritis but has no effect on the underlying inflammation, redness, and swelling of the joint. It is as effective as the non-steroidal anti-inflammatory
drug ibuprofen
in relieving the pain of osteoarthritis of the knee.
Paracetamol has relatively little anti-inflammatory activity, unlike other common analgesics such as the NSAID
s aspirin and ibuprofen.
Regarding comparative efficacy, studies show conflicting results when compared to NSAIDs. A randomized controlled trial
of chronic pain from osteoarthritis in adults found similar benefit from paracetamol and ibuprofen.
The efficacy of paracetamol when used in a combination form with weak opioids (such as codeine) has been questioned by recent data studies; the small amount of data available have made reaching a conclusion difficult. Combination drugs of paracetamol and strong opioids like morphine have been shown to reduce the amount of opioid used and improve analgesic effect.
A randomized controlled trial of acute musculoskeletal pain in children found that the standard over-the-counter dose of ibuprofen gives greater pain relief than the standard dose of paracetamol.
s of paracetamol are mild to non-existent. In contrast to aspirin, it is not a blood thinner
(and thus may be used in patients where coagulation is a concern), and it does not cause gastric irritation. Compared to Ibuprofen—which can have adverse effects that include diarrhea, vomiting, and abdominal pain—acetaminophen is well tolerated with fewer side effects. Prolonged daily use increases the risk of upper gastrointestinal complications such as stomach bleeding
, and may cause kidney or liver damage. Paracetamol is metabolized by the liver and is hepatotoxic; side effects may be more likely in chronic alcoholics
or patients with liver damage.
Until 2010 paracetamol was believed safe in pregnancy (as it does not affect the closure of the fetal ductus arteriosus
as NSAIDs can). However, in a study published in October 2010 it has been linked to infertility
in the adult life of the unborn. Like NSAIDs and unlike opioid analgesics, paracetamol has not been found to cause euphoria or alter mood although recent research shows some evidence that paracetamol can ease psychological pain. Unlike aspirin, it is safe for children, as paracetamol is not associated with a risk of Reye's syndrome
in children with viral illnesses. Paracetamol use for fever in the first year of life was associated with an increase in the incidence of asthma
tic symptoms at 6–7 years, and that paracetamol use, both in the first year of life and in children aged 6–7 years, was associated with an increased incidence of rhinoconjunctivitis
and eczema
. The authors acknowledged that their "findings might have been due to confounding by indication", i. e., that the association may not be causal but rather due to the disease being treated with paracetamol, and emphasized that further research is needed. Furthermore a number of editorials, comments, correspondence, and their replies have been published in The Lancet concerning the methodology and conclusions of this study. The UK regulatory body the Medicines and Healthcare products Regulatory Agency
, also reviewed this research and published a number of concerns over data interpretation, and offer the following advice for healthcare professionals, parents, and care-givers: "The results of this new study do not necessitate any change to the current guidance for use in children. Paracetamol remains a safe and appropriate choice of analgesic in children. There is insufficient evidence from this research to change guidance regarding the use of antipyretic
s in children. "
Chronic users of acetaminophen may have a higher risk of developing blood cancer.
s in the US than overdose of any other pharmacological substance. Signs and symptoms of paracetamol toxicity may initially be absent or vague. Untreated overdose can lead to liver failure
and death within days. Treatment is aimed at removing the paracetamol from the body and replacing glutathione
. Activated charcoal can be used to decrease absorption of paracetamol if the patient presents for treatment soon after the overdose. While the antidote, acetylcysteine, (also called N-acetylcysteine or NAC) acts as a precursor for glutathione, helping the body regenerate enough to prevent damage to the liver, a liver transplant is often required if damage to the liver becomes severe.
There are tablets available (brand-name in the UK Paradote) that combine paracetamol with an antidote (methionine
), to protect the liver in case of an overdose.
In June 2009, a U. S. Food and Drug Administration (FDA) advisory committee recommended that new restrictions should be placed on paracetamol usage in the United States to help protect people from the potential toxic effects. The maximum dosage at any given time would be decreased from 1000 mg to 650 mg, while combinations of paracetamol and narcotic
analgesic
s would be prohibited. Committee members were particularly concerned by the fact that the present maximum dosages of paracetamol had been shown to produce alterations in hepatic
function. On January 13, 2011, the FDA asked manufacturers of prescription combination products containing paracetamol to limit the amount of paracetamol to no more than 325 mg per tablet or capsule and began requiring manufacturers to update the labels of these products to warn of the potential risk for severe liver damage. Manufacturers will have three years to limit the amount of paracetamol in their prescription drug products to 325 mg per dosage unit. The FDA also is requiring manufacturers to update labels of all prescription combination paracetamol products to warn of the potential risk for severe liver injury.
In November 2011, the Medicines and Healthcare products Regulatory Agency
revised UK dosing of liquid paracetamol for children.
analgesics"; it is the only such drug still in use today. It is classified as a nonsteroidal anti-inflammatory drug (NSAID) by some sources, and not as an NSAID by others, while most sources implicitly distinguish them, for example by mentioning both NSAIDs and paracetamol in the same sentence. Paracetamol has few anti-inflammatory effects in comparison to NSAIDs. However, aspirin
, paracetamol and other NSAIDs all act by the same mechanism (inhibition of prostaglandin
synthesis) and all show varying levels of analgesic, anti-inflammatory, antipyretic and antiplatelet actions.
 To date, the mechanism of action of paracetamol is not completely understood. The main mechanism proposed is the inhibition of cyclooxygenase
To date, the mechanism of action of paracetamol is not completely understood. The main mechanism proposed is the inhibition of cyclooxygenase
(COX), and recent findings suggest that it is highly selective for COX-2. While it has analgesic
and antipyretic
properties comparable to those of aspirin
or other NSAIDs, its peripheral anti-inflammatory activity is usually limited by several factors, one of which is high level of peroxides present in inflammatory
lesions. However, in some circumstances, even peripheral anti-inflammatory activity comparable to other NSAIDs can be observed. An article in nature communications from a research group in Lund, Sweden in November 2011 has found a hint to the analgesic mechanism of acetaminophen (paracetamol), being that the metabolites of acetaminophen e.g NAPQI, act on TRPA1-receptors in the spinal cord to suppress the signal transduction from the superficial layers of the dorsal horn, to alleviate pain.
Because of its selectivity for COX-2 it does not significantly inhibit the production of the pro-clotting thromboxane
s.
The COX family of enzymes are responsible for the metabolism of arachidonic acid
to prostaglandin H2
, an unstable molecule that is, in turn, converted to numerous other pro-inflammatory compounds. Classical anti-inflammatories such as the NSAIDs block this step. Only when appropriately oxidized is the COX enzyme highly active.
Paracetamol reduces the oxidized form of the COX enzyme, preventing it from forming pro-inflammatory chemicals. This leads to a reduced amount of Prostaglandin E2 in the CNS, thus lowering the hypothalamic set-point in the thermoregulatory centre.
Paracetamol also modulates the endogenous cannabinoid system
. Paracetamol is metabolized to AM404
, a compound with several actions; what is most important is that it inhibits the uptake of the endogenous cannabinoid/vanilloid anandamide
by neurons. Anandamide uptake would result in the activation of the main pain receptor (nociceptor) of the body, the TRPV1
(older name: vanilloid receptor). Furthermore, AM404 inhibits sodium channels, as do the anesthetics lidocaine and procaine. Either of these actions by themselves has been shown to reduce pain, and are a possible mechanism for paracetamol. However, it has been demonstrated that, after blocking cannabinoid receptors with synthetic antagonists, paracetamol's analgesic effects are prevented, suggesting its pain-relieving action involves the endogenous cannabinoid system. Spinal TRPA1
receptors have also been demonstrated to mediate antinociceptive effects of paracetamol and Δ9-tetrahydrocannabiorcol in mice.
The exact mechanisms by which COX is inhibited in various circumstances is still a subject of discussion. Because of differences in the activity of paracetamol, aspirin, and other NSAIDs, it has been postulated that further COX variants may exist. A recently discovered COX-1 splice variant termed COX-3
was considered to explain some of the knowledge gap but newer findings do not support the hypothesis that it plays any significant role in the functioning of paracetamol.
Aspirin is known to inhibit the cyclooxygenase
(COX) family of enzymes and, because paracetamol's action is partially similar to aspirin's, much research has focused on whether paracetamol also inhibits COX. It is now clear that paracetamol acts via at least two pathways.
One theory holds that paracetamol works by inhibiting the COX-3
isoform of the COX family of enzymes. When expressed in dogs, this enzyme shares a strong similarity to the other COX enzymes, produces pro-inflammatory chemicals, and is selectively inhibited by paracetamol. However, some research has suggested that, in humans and mice, the COX-3 enzyme is without inflammatory action. Another possibility is that paracetamol blocks cyclooxygenase (as in aspirin), but that is in an inflammatory environment where the concentration of peroxides is high, and the high oxidation state of paracetamol prevents its actions. This would mean that paracetamol has no direct effect at the site of inflammation, but instead acts in the CNS where the environment is not oxidative, to reduce temperature, etc. The exact mechanism by which paracetamol is believed to affect COX-3 is disputed.
 Paracetamol consists of a benzene
Paracetamol consists of a benzene
ring core, substituted
by one hydroxyl
group and the nitrogen
atom of an amide
group in the para (1,4) pattern
. The amide group is acetamide
(ethanamide). It is an extensively conjugated system
, as the lone pair
on the hydroxyl oxygen, the benzene pi cloud, the nitrogen lone pair, the p orbital
on the carbonyl
carbon, and the lone pair on the carbonyl oxygen are all conjugated. The presence of two activating groups also make the benzene ring highly reactive toward electrophilic
aromatic substitution. As the substituents are ortho,para-directing and para with respect to each other, all positions on the ring are more or less equally activated. The conjugation also greatly reduces the basicity
of the oxygens and the nitrogen, while making the hydroxyl acidic through delocalisation of charge developed on the phenoxide
anion
.
phenol with sodium nitrate
, separating the desired p-nitrophenol
from the ortho- byproduct, and reducing the nitro group with sodium borohydride
. The resultant p-aminophenol is then acetylated with acetic anhydride
. In this reaction, phenol
is strongly activating, thus the reaction requires only mild conditions (cf. the nitration of benzene). The industrial process is analogous, but hydrogenation is used instead of the sodium borohydride reduction.
A simpler synthesis by Hoechst-Celanese involves direct acylation of phenol with acetic anhydride catalyzed by HF, conversion of the ketone to a ketoxime with hydroxylamine
, followed by the acid-catalyzed Beckmann rearrangement
to give the amide.
Demand for paracetamol in the United States was estimated at 30–35 thousand tonnes per year in 1997, equal to the demand from the rest of the world.
 Paracetamol is metabolised
Paracetamol is metabolised
primarily in the liver
, into non-toxic products. Three metabolic pathway
s are notable:
All three pathways yield final products that are inactive, non-toxic, and eventually excreted by the kidneys. In the third pathway, however, the intermediate product NAPQI is toxic. NAPQI is primarily responsible for the toxic effects of paracetamol; this constitutes an example of toxication
.
Production of NAPQI is due primarily to two isoenzymes of cytochrome P450: CYP2E1
and CYP1A2
. The P450 gene is highly polymorphic
, however, and individual differences in paracetamol toxicity are believed due to a third isoenzyme, CYP2D6
. Genetic polymorphisms in CYP2D6
may contribute to significantly different rates of production of NAPQI. Furthermore, individuals can be classified as "extensive", "ultrarapid", and "poor" metabolizers (producers of NAPQI), depending on their levels of CYP2D6 expression. Although CYP2D6 metabolises paracetamol into NAPQI to a lesser extent than other P450 enzymes, its activity may contribute to paracetamol toxicity in extensive and ultrarapid metabolisers, and when paracetamol is taken at very large doses. At usual doses, NAPQI is quickly detoxified by conjugation. Following overdose, and possibly also in extensive and ultrarapid metabolizers, this detoxification pathway becomes saturated, and, as a consequence, NAPQI accumulates.
of paracetamol. p-Aminophenol prepared this way, and related to the commercially available Metol
, has been used as a developer in photography by hobbyists. This reaction is also used to determine paracetamol in urine samples: After hydrolysis with hydrochloric acid, p-aminophenol reacts in ammonia solution with a phenol derivate, e.g. salicylic acid, to form an indophenol
dye under oxidization by air.
 Acetanilide was the first aniline derivative serendipitously found to possess analgesic as well as antipyretic properties, and was quickly introduced into medical practice under the name of Antifebrin by A. Cahn and P. Hepp in 1886. But its unacceptable toxic effects, the most alarming being cyanosis
Acetanilide was the first aniline derivative serendipitously found to possess analgesic as well as antipyretic properties, and was quickly introduced into medical practice under the name of Antifebrin by A. Cahn and P. Hepp in 1886. But its unacceptable toxic effects, the most alarming being cyanosis
due to methemoglobinemia
, prompted the search for less toxic aniline derivatives. Harmon Northrop Morse
had already synthesized paracetamol at Johns Hopkins University
via the reduction of p-nitrophenol
with tin
in glacial acetic acid
in 1877,
but it was not until 1887 that clinical pharmacologist Joseph von Mering
tried paracetamol on patients. In 1893, von Mering published a paper reporting on the clinical results of paracetamol with phenacetin
, another aniline derivative. Von Mering claimed that, unlike phenacetin, paracetamol had a slight tendency to produce methemoglobinemia. Paracetamol was then quickly discarded in favor of phenacetin. The sales of phenacetin established Bayer
as a leading pharmaceutical company. Overshadowed in part by aspirin
, introduced into medicine by Heinrich Dreser
in 1899, phenacetin was popular for many decades, particularly in widely advertised over-the-counter "headache mixtures", usually containing phenacetin, an aminopyrine derivative of aspirin, caffeine, and sometimes a barbiturate
.
Von Mering's claims remained essentially unchallenged for half a century, until two teams of researchers from the United States analyzed the metabolism of acetanilide and paracetamol. In 1947 David Lester
and Leon Greenberg found strong evidence that paracetamol was a major metabolite of acetanilide in human blood, and in a subsequent study they reported that large doses of paracetamol given to albino rats did not cause methemoglobinemia. In three papers published in the September 1948 issue of the Journal of Pharmacology and Experimental Therapeutics
, Bernard Brodie
, Julius Axelrod
and Frederick Flinn confirmed using more specific methods that paracetamol was the major metabolite of acetanilide in human blood, and established it was just as efficacious an analgesic as its precursor. They also suggested that methemoglobinemia is produced in humans mainly by another metabolite, phenylhydroxylamine
. A followup paper by Brodie and Axelrod in 1949 established that phenacetin was also metabolized to paracetamol. This led to a "rediscovery" of paracetamol. It has been suggested that contamination of paracetamol with 4-aminophenol, the substance von Mering synthesized it from, may be the cause for his spurious findings.
Paracetamol was first marketed in the United States in 1953 by Sterling-Winthrop Co., which promoted it as preferable to aspirin since it was safe to take for children and people with ulcers. The best known brand today for paracetamol in the United States, Tylenol
, was established in 1955 when McNeil Laboratories
started selling paracetamol as a pain and fever reliever for children, under the brand name Tylenol Children's Elixir—the word "tylenol" was a contraction of para-acetylaminophenol. In 1956, 500 mg tablets of paracetamol went on sale in the United Kingdom under the trade name Panadol, produced by Frederick Stearns & Co, a subsidiary of Sterling Drug
Inc. Panadol was originally available only by prescription, for the relief of pain and fever, and was advertised as being "gentle to the stomach," since other analgesic agents of the time contained aspirin, a known stomach irritant. In 1963, paracetamol was added to the British Pharmacopoeia
, and has gained popularity since then as an analgesic agent with few side-effects and little interaction with other pharmaceutical agents. Concerns about paracetamol's safety delayed its widespread acceptance until the 1970s, but in the 1980s paracetamol sales exceeded those of aspirin in many countries, including the United Kingdom. This was accompanied by the commercial demise of phenacetin, blamed as the cause of analgesic nephropathy and hematological toxicity.
The U.S. patent
on paracetamol has long expired, and generic versions of the drug are widely available under the Drug Price Competition and Patent Term Restoration Act
of 1984, although certain Tylenol preparations were protected until 2007. U.S. patent 6,126,967 filed September 3, 1998 was granted for "Extended release acetaminophen particles".
, capsule, liquid suspension, suppository
, intravenous, and intramuscular form. The common adult dose is 500 mg to 1000 mg. The recommended maximum daily dose, for adults, is 4000 mg. In recommended doses, paracetamol generally is safe for children and infants, as well as for adults, although rare cases of acute liver injury have been linked to amounts lower than 2500 mg per day.
Panadol
, which is marketed in Africa, Asia, Europe, Central America, and Australasia
, is the most widely available brand of paracetamol, sold in over 80 countries. In North America, paracetamol is sold in generic form (usually labeled as acetaminophen) or under a number of trade names, for instance, Tylenol
(McNeil-PPC, Inc.
), Anacin-3
, Tempra, Datril, and Ofirmev. While there is brand named paracetamol available in the UK (e.g. Panadol), unbranded or generic paracetamol is more commonly sold. Acamol
, a brand name for paracetamol produced by Teva Pharmaceutical Industries
in Israel
, is one of the most widely used drugs in that country. In the Philippines, the largest-selling paracetamol brand is Biogesic, manufactured by the drug giant United Laboratories. Biogesic tablet sales reach nearly a billion units each year in the country alone, not including liquid suspension formats. The brand is also available in most of the ASEAN countries where the drug giant has market presence.
In Europe, the most common brands of paracetamol are Efferalgan and Doliprane. In India, the most common brand of paracetamol is Crocin manufactured by Glaxo SmithKline Asia. In Bangladesh the most popular brand is Napa manufactured by Beximco Pharma. In China paracetamol is sold over the counter as Duìyǐxiān'ānjīfēn Piàn (对乙酰氨基酚片). Likewise in Japan it is sold under the name Acetaminophen (アセトアミノフェン Asetoaminofen). In North Korea the DPRK-Swiss joint venture PyongSu Pharma
markets the drug as PyongSu Cetamol.
In some formulations, paracetamol is combined with the opioid
codeine
, sometimes referred to as co-codamol
(BAN
). In the United States and Canada, this is marketed under the name of Tylenol #1/2/3/4, which contain 8–10 mg, 15 mg, 30 mg, and 60 mg of codeine
, respectively. In the U.S., this combination is available only by prescription, while the lowest-strength preparation is over-the-counter in Canada, and, in other countries, other strengths may be available over the counter. There are generic forms of these combinations as well. In the UK and in many other countries, this combination is marketed under the names of Tylex CD and Panadeine. Other names include Captin, Disprol, Dymadon, Fensum, Hedex, Mexalen, Nofedol, Paralen, Pediapirin, Perfalgan, and Solpadeine. Paracetamol is also combined with other opioids such as dihydrocodeine
, referred to as co-dydramol
(BAN
), oxycodone
or hydrocodone
, marketed in the U.S. as Percocet
and Vicodin
, respectively. Another very commonly used analgesic combination includes paracetamol in combination with propoxyphene napsylate, sold under the brand name Darvocet. A combination of paracetamol, codeine, and the calmative doxylamine succinate
is marketed as Syndol or Mersyndol. The efficacy of paracetamol/codeine combinations have been questioned by recent research.
Paracetamol is commonly used in multi-ingredient preparations for migraine
headache, typically including butalbital
and paracetamol with or without caffeine
, and sometimes containing codeine.
). Treatment with N-acetylcysteine, methylene blue
or both is sometimes effective after the ingestion of small doses of paracetamol.
Although paracetamol is believed to have no significant anti-inflammatory activity, it has been reported as effective as aspirin in the treatment of musculoskeletal pain in dogs. A paracetamol-codeine product (trade name Pardale-V) licensed for use in dogs is available on veterinary prescription in the UK. It should be administered to dogs only on veterinary advice and with extreme caution. The main effects of toxicity in dogs is liver damage, GI ulceration has been reported. N-acetylcysteine treatment is efficacious in dogs when administered within a few hours of paracetamol ingestion.
Paracetamol is also lethal to snakes, and has been suggested as a chemical control program for the invasive brown tree snake
(Boiga irregularis) in Guam
.
International Nonproprietary Name
An International Nonproprietary Name is the official nonproprietary or generic name given to a pharmaceutical substance, as designated by the World Health Organization...
(icon or ˌ), or acetaminophen USAN
United States Adopted Name
United States Adopted Names are unique nonproprietary names assigned to pharmaceuticals marketed in the United States. Each name is assigned by the USAN Council, which is co-sponsored by the American Medical Association , the United States Pharmacopeial Convention , and the American Pharmacists...
, is a widely used over-the-counter
Over-the-counter drug
Over-the-counter drugs are medicines that may be sold directly to a consumer without a prescription from a healthcare professional, as compared to prescription drugs, which may be sold only to consumers possessing a valid prescription...
analgesic
Analgesic
An analgesic is any member of the group of drugs used to relieve pain . The word analgesic derives from Greek an- and algos ....
(pain reliever) and antipyretic
Antipyretic
Antipyretics ; an-tee-pahy-ret-iks; from the Greek anti, against, and pyreticus, are drugs or herbs that reduce fever. Normally, they will not lower body temperature if one does not have a fever. Antipyretics cause the hypothalamus to override an interleukin-induced increase in temperature...
(fever reducer). It is commonly used for the relief of headache
Headache
A headache or cephalalgia is pain anywhere in the region of the head or neck. It can be a symptom of a number of different conditions of the head and neck. The brain tissue itself is not sensitive to pain because it lacks pain receptors. Rather, the pain is caused by disturbance of the...
s and other minor aches and pains and is a major ingredient in numerous cold
Common cold
The common cold is a viral infectious disease of the upper respiratory system, caused primarily by rhinoviruses and coronaviruses. Common symptoms include a cough, sore throat, runny nose, and fever...
and flu
Influenza
Influenza, commonly referred to as the flu, is an infectious disease caused by RNA viruses of the family Orthomyxoviridae , that affects birds and mammals...
remedies. In combination with opioid analgesics, paracetamol can also be used in the management of more severe pain such as post surgical pain and providing palliative care
Palliative care
Palliative care is a specialized area of healthcare that focuses on relieving and preventing the suffering of patients...
in advanced cancer patients. The onset of analgesia is approximately 11 minutes after oral administration
Oral administration
Oral administration is a route of administration where a substance is taken through the mouth.-Terminology:Per os is an adverbial phrase meaning literally from Latin "by mouth" or "by way of the mouth." The expression is used in medicine to describe a treatment that is taken orally. The...
of paracetamol, and its half-life
Biological half-life
The biological half-life or elimination half-life of a substance is the time it takes for a substance to lose half of its pharmacologic, physiologic, or radiologic activity, as per the MeSH definition...
is 1–4 hours.
While generally safe for use at recommended doses ( and up to for adults), acute overdoses of paracetamol can cause potentially fatal liver damage
Hepatotoxicity
Hepatotoxicity implies chemical-driven liver damage.The liver plays a central role in transforming and clearing chemicals and is susceptible to the toxicity from these agents. Certain medicinal agents, when taken in overdoses and sometimes even when introduced within therapeutic ranges, may injure...
and, in rare individuals, a normal dose can do the same; the risk is heightened by alcohol consumption
Ethanol
Ethanol, also called ethyl alcohol, pure alcohol, grain alcohol, or drinking alcohol, is a volatile, flammable, colorless liquid. It is a psychoactive drug and one of the oldest recreational drugs. Best known as the type of alcohol found in alcoholic beverages, it is also used in thermometers, as a...
. Paracetamol toxicity
Paracetamol toxicity
Paracetamol toxicity is caused by excessive use or overdose of the analgesic drug paracetamol . Mainly causing liver injury, paracetamol toxicity is one of the most common causes of poisoning worldwide...
is the foremost cause of acute liver failure in the Western world
Western world
The Western world, also known as the West and the Occident , is a term referring to the countries of Western Europe , the countries of the Americas, as well all countries of Northern and Central Europe, Australia and New Zealand...
, and accounts for most drug overdoses in the United States, the United Kingdom, Australia and New Zealand.
It is the active metabolite of phenacetin
Phenacetin
Phenacetin is an analgesic, once widely used; its use has declined because of its adverse effects.-History:Phenacetin was introduced in 1887, and was used principally as an analgesic, and was one of the first synthetic fever reducers to go on the market...
, once popular as an analgesic and antipyretic in its own right, but unlike phenacetin and its combinations, paracetamol is not considered carcinogen
Carcinogen
A carcinogen is any substance, radionuclide, or radiation that is an agent directly involved in causing cancer. This may be due to the ability to damage the genome or to the disruption of cellular metabolic processes...
ic at therapeutic doses. The words acetaminophen (used in the United States, Canada, Japan, South Korea, Hong Kong, and Iran) and paracetamol (used elsewhere) both come from a chemical name for the compound: para-acetylaminophenol and para-acetylaminophenol. In some contexts, it is simply abbreviated as APAP, for acetyl-para-aminophenol.
Fever
Paracetamol is approved for reducing feverFever
Fever is a common medical sign characterized by an elevation of temperature above the normal range of due to an increase in the body temperature regulatory set-point. This increase in set-point triggers increased muscle tone and shivering.As a person's temperature increases, there is, in...
in people of all ages. The World Health Organization
World Health Organization
The World Health Organization is a specialized agency of the United Nations that acts as a coordinating authority on international public health. Established on 7 April 1948, with headquarters in Geneva, Switzerland, the agency inherited the mandate and resources of its predecessor, the Health...
(WHO) recommends that paracetamol only be used to treat fever in children if their temperature is greater than 38.5 °C (101.3 °F). The efficacy of paracetamol by itself in children with fevers has been questioned and a meta-analysis showed that it is less effective than ibuprofen
Ibuprofen
Ibuprofen is a nonsteroidal anti-inflammatory drug used for relief of symptoms of arthritis, fever, as an analgesic , especially where there is an inflammatory component, and dysmenorrhea....
.
Paracetamol has a well-established role in pediatric medicine as an effective analgesic and antipyretic.
Pain
Paracetamol is used for the relief of pains associated with many parts of the body. It has analgesicAnalgesic
An analgesic is any member of the group of drugs used to relieve pain . The word analgesic derives from Greek an- and algos ....
properties comparable to those of aspirin
Aspirin
Aspirin , also known as acetylsalicylic acid , is a salicylate drug, often used as an analgesic to relieve minor aches and pains, as an antipyretic to reduce fever, and as an anti-inflammatory medication. It was discovered by Arthur Eichengrun, a chemist with the German company Bayer...
, while its anti-inflammatory effects are weaker. It is better tolerated than aspirin in patients in whom excessive gastric acid
Gastric acid
Gastric acid is a digestive fluid, formed in the stomach. It has a pH of 1 to 2 and is composed of hydrochloric acid , and large quantities of potassium chloride and sodium chloride...
secretion or prolongation of bleeding time may be a concern. Available without a prescription, it has in recent years increasingly become a common household drug.
Paracetamol can relieve pain in mild arthritis but has no effect on the underlying inflammation, redness, and swelling of the joint. It is as effective as the non-steroidal anti-inflammatory
Anti-inflammatory
Anti-inflammatory refers to the property of a substance or treatment that reduces inflammation. Anti-inflammatory drugs make up about half of analgesics, remedying pain by reducing inflammation as opposed to opioids, which affect the central nervous system....
drug ibuprofen
Ibuprofen
Ibuprofen is a nonsteroidal anti-inflammatory drug used for relief of symptoms of arthritis, fever, as an analgesic , especially where there is an inflammatory component, and dysmenorrhea....
in relieving the pain of osteoarthritis of the knee.
Paracetamol has relatively little anti-inflammatory activity, unlike other common analgesics such as the NSAID
Non-steroidal anti-inflammatory drug
Nonsteroidal anti-inflammatory drugs, usually abbreviated to NSAIDs or NAIDs, but also referred to as nonsteroidal anti-inflammatory agents/analgesics or nonsteroidal Anti-inflammatory medicines , are drugs with analgesic and antipyretic effects and which have, in higher doses, anti-inflammatory...
s aspirin and ibuprofen.
Regarding comparative efficacy, studies show conflicting results when compared to NSAIDs. A randomized controlled trial
Randomized controlled trial
A randomized controlled trial is a type of scientific experiment - a form of clinical trial - most commonly used in testing the safety and efficacy or effectiveness of healthcare services or health technologies A randomized controlled trial (RCT) is a type of scientific experiment - a form of...
of chronic pain from osteoarthritis in adults found similar benefit from paracetamol and ibuprofen.
The efficacy of paracetamol when used in a combination form with weak opioids (such as codeine) has been questioned by recent data studies; the small amount of data available have made reaching a conclusion difficult. Combination drugs of paracetamol and strong opioids like morphine have been shown to reduce the amount of opioid used and improve analgesic effect.
A randomized controlled trial of acute musculoskeletal pain in children found that the standard over-the-counter dose of ibuprofen gives greater pain relief than the standard dose of paracetamol.
Adverse effects
In recommended doses, the side effectSide effect
In medicine, a side effect is an effect, whether therapeutic or adverse, that is secondary to the one intended; although the term is predominantly employed to describe adverse effects, it can also apply to beneficial, but unintended, consequences of the use of a drug.Occasionally, drugs are...
s of paracetamol are mild to non-existent. In contrast to aspirin, it is not a blood thinner
Anticoagulant
An anticoagulant is a substance that prevents coagulation of blood. A group of pharmaceuticals called anticoagulants can be used in vivo as a medication for thrombotic disorders. Some anticoagulants are used in medical equipment, such as test tubes, blood transfusion bags, and renal dialysis...
(and thus may be used in patients where coagulation is a concern), and it does not cause gastric irritation. Compared to Ibuprofen—which can have adverse effects that include diarrhea, vomiting, and abdominal pain—acetaminophen is well tolerated with fewer side effects. Prolonged daily use increases the risk of upper gastrointestinal complications such as stomach bleeding
Upper gastrointestinal bleeding
Upper gastrointestinal bleeding refers to hemorrhage in the upper gastrointestinal tract. The anatomic cut-off for upper GI bleeding is the ligament of Treitz, which connects the fourth portion of the duodenum to the diaphragm near the splenic flexure of the colon.Upper GI bleeds are considered...
, and may cause kidney or liver damage. Paracetamol is metabolized by the liver and is hepatotoxic; side effects may be more likely in chronic alcoholics
Alcoholism
Alcoholism is a broad term for problems with alcohol, and is generally used to mean compulsive and uncontrolled consumption of alcoholic beverages, usually to the detriment of the drinker's health, personal relationships, and social standing...
or patients with liver damage.
Until 2010 paracetamol was believed safe in pregnancy (as it does not affect the closure of the fetal ductus arteriosus
Ductus arteriosus
In the developing fetus, the ductus arteriosus , also called the ductus Botalli, is a shunt connecting the pulmonary artery to the aortic arch. It allows most of the blood from the right ventricle to bypass the fetus's fluid-filled lungs. Upon closure at birth, it becomes the ligamentum arteriosum...
as NSAIDs can). However, in a study published in October 2010 it has been linked to infertility
Infertility
Infertility primarily refers to the biological inability of a person to contribute to conception. Infertility may also refer to the state of a woman who is unable to carry a pregnancy to full term...
in the adult life of the unborn. Like NSAIDs and unlike opioid analgesics, paracetamol has not been found to cause euphoria or alter mood although recent research shows some evidence that paracetamol can ease psychological pain. Unlike aspirin, it is safe for children, as paracetamol is not associated with a risk of Reye's syndrome
Reye's syndrome
Reye's syndrome is a potentially fatal disease that causes numerous detrimental effects to many organs, especially the brain and liver, as well as causing a lower than usual level of blood sugar . The classic features are liver damage, aspirin use and a viral infection...
in children with viral illnesses. Paracetamol use for fever in the first year of life was associated with an increase in the incidence of asthma
Asthma
Asthma is the common chronic inflammatory disease of the airways characterized by variable and recurring symptoms, reversible airflow obstruction, and bronchospasm. Symptoms include wheezing, coughing, chest tightness, and shortness of breath...
tic symptoms at 6–7 years, and that paracetamol use, both in the first year of life and in children aged 6–7 years, was associated with an increased incidence of rhinoconjunctivitis
Allergic conjunctivitis
Allergic conjunctivitis is inflammation of the conjunctiva due to allergy. Although allergens differ between patients, the most common cause is hay fever. Symptoms consist of redness , oedema of the conjunctiva, itching and increased lacrimation...
and eczema
Eczema
Eczema is a form of dermatitis, or inflammation of the epidermis . In England, an estimated 5.7 million or about one in every nine people have been diagnosed with the disease by a clinician at some point in their lives.The term eczema is broadly applied to a range of persistent skin conditions...
. The authors acknowledged that their "findings might have been due to confounding by indication", i. e., that the association may not be causal but rather due to the disease being treated with paracetamol, and emphasized that further research is needed. Furthermore a number of editorials, comments, correspondence, and their replies have been published in The Lancet concerning the methodology and conclusions of this study. The UK regulatory body the Medicines and Healthcare products Regulatory Agency
Medicines and Healthcare products Regulatory Agency
The Medicines and Healthcare products Regulatory Agency is the UK government agency which is responsible for ensuring that medicines and medical devices work and are acceptably safe....
, also reviewed this research and published a number of concerns over data interpretation, and offer the following advice for healthcare professionals, parents, and care-givers: "The results of this new study do not necessitate any change to the current guidance for use in children. Paracetamol remains a safe and appropriate choice of analgesic in children. There is insufficient evidence from this research to change guidance regarding the use of antipyretic
Antipyretic
Antipyretics ; an-tee-pahy-ret-iks; from the Greek anti, against, and pyreticus, are drugs or herbs that reduce fever. Normally, they will not lower body temperature if one does not have a fever. Antipyretics cause the hypothalamus to override an interleukin-induced increase in temperature...
s in children. "
Chronic users of acetaminophen may have a higher risk of developing blood cancer.
Overdose
Paracetamol hepatotoxicity is, by far, the most common cause of acute liver failure in both the United States and the United Kingdom. Paracetamol overdose results in more calls to poison control centerPoison control center
A poison control center is a medical facility that is able to provide immediate, free, and expert treatment advice and assistance over the telephone in case of exposure to poisonous or hazardous substances...
s in the US than overdose of any other pharmacological substance. Signs and symptoms of paracetamol toxicity may initially be absent or vague. Untreated overdose can lead to liver failure
Liver failure
Acute liver failure is the appearance of severe complications rapidly after the first signs of liver disease , and indicates that the liver has sustained severe damage . The complications are hepatic encephalopathy and impaired protein synthesis...
and death within days. Treatment is aimed at removing the paracetamol from the body and replacing glutathione
Glutathione
Glutathione is a tripeptide that contains an unusual peptide linkage between the amine group of cysteine and the carboxyl group of the glutamate side-chain...
. Activated charcoal can be used to decrease absorption of paracetamol if the patient presents for treatment soon after the overdose. While the antidote, acetylcysteine, (also called N-acetylcysteine or NAC) acts as a precursor for glutathione, helping the body regenerate enough to prevent damage to the liver, a liver transplant is often required if damage to the liver becomes severe.
There are tablets available (brand-name in the UK Paradote) that combine paracetamol with an antidote (methionine
Methionine
Methionine is an α-amino acid with the chemical formula HO2CCHCH2CH2SCH3. This essential amino acid is classified as nonpolar. This amino-acid is coded by the codon AUG, also known as the initiation codon, since it indicates mRNA's coding region where translation into protein...
), to protect the liver in case of an overdose.
In June 2009, a U. S. Food and Drug Administration (FDA) advisory committee recommended that new restrictions should be placed on paracetamol usage in the United States to help protect people from the potential toxic effects. The maximum dosage at any given time would be decreased from 1000 mg to 650 mg, while combinations of paracetamol and narcotic
Narcotic
The term narcotic originally referred medically to any psychoactive compound with any sleep-inducing properties. In the United States of America it has since become associated with opioids, commonly morphine and heroin and their derivatives, such as hydrocodone. The term is, today, imprecisely...
analgesic
Analgesic
An analgesic is any member of the group of drugs used to relieve pain . The word analgesic derives from Greek an- and algos ....
s would be prohibited. Committee members were particularly concerned by the fact that the present maximum dosages of paracetamol had been shown to produce alterations in hepatic
Liver
The liver is a vital organ present in vertebrates and some other animals. It has a wide range of functions, including detoxification, protein synthesis, and production of biochemicals necessary for digestion...
function. On January 13, 2011, the FDA asked manufacturers of prescription combination products containing paracetamol to limit the amount of paracetamol to no more than 325 mg per tablet or capsule and began requiring manufacturers to update the labels of these products to warn of the potential risk for severe liver damage. Manufacturers will have three years to limit the amount of paracetamol in their prescription drug products to 325 mg per dosage unit. The FDA also is requiring manufacturers to update labels of all prescription combination paracetamol products to warn of the potential risk for severe liver injury.
In November 2011, the Medicines and Healthcare products Regulatory Agency
Medicines and Healthcare products Regulatory Agency
The Medicines and Healthcare products Regulatory Agency is the UK government agency which is responsible for ensuring that medicines and medical devices work and are acceptably safe....
revised UK dosing of liquid paracetamol for children.
Classification
Paracetamol is part of the class of drugs known as "anilineAniline
Aniline, phenylamine or aminobenzene is an organic compound with the formula C6H5NH2. Consisting of a phenyl group attached to an amino group, aniline is the prototypical aromatic amine. Being a precursor to many industrial chemicals, its main use is in the manufacture of precursors to polyurethane...
analgesics"; it is the only such drug still in use today. It is classified as a nonsteroidal anti-inflammatory drug (NSAID) by some sources, and not as an NSAID by others, while most sources implicitly distinguish them, for example by mentioning both NSAIDs and paracetamol in the same sentence. Paracetamol has few anti-inflammatory effects in comparison to NSAIDs. However, aspirin
Aspirin
Aspirin , also known as acetylsalicylic acid , is a salicylate drug, often used as an analgesic to relieve minor aches and pains, as an antipyretic to reduce fever, and as an anti-inflammatory medication. It was discovered by Arthur Eichengrun, a chemist with the German company Bayer...
, paracetamol and other NSAIDs all act by the same mechanism (inhibition of prostaglandin
Prostaglandin
A prostaglandin is any member of a group of lipid compounds that are derived enzymatically from fatty acids and have important functions in the animal body. Every prostaglandin contains 20 carbon atoms, including a 5-carbon ring....
synthesis) and all show varying levels of analgesic, anti-inflammatory, antipyretic and antiplatelet actions.
Mechanism of action
Cyclooxygenase
Cyclooxygenase is an enzyme that is responsible for formation of important biological mediators called prostanoids, including prostaglandins, prostacyclin and thromboxane. Pharmacological inhibition of COX can provide relief from the symptoms of inflammation and pain...
(COX), and recent findings suggest that it is highly selective for COX-2. While it has analgesic
Analgesic
An analgesic is any member of the group of drugs used to relieve pain . The word analgesic derives from Greek an- and algos ....
and antipyretic
Antipyretic
Antipyretics ; an-tee-pahy-ret-iks; from the Greek anti, against, and pyreticus, are drugs or herbs that reduce fever. Normally, they will not lower body temperature if one does not have a fever. Antipyretics cause the hypothalamus to override an interleukin-induced increase in temperature...
properties comparable to those of aspirin
Aspirin
Aspirin , also known as acetylsalicylic acid , is a salicylate drug, often used as an analgesic to relieve minor aches and pains, as an antipyretic to reduce fever, and as an anti-inflammatory medication. It was discovered by Arthur Eichengrun, a chemist with the German company Bayer...
or other NSAIDs, its peripheral anti-inflammatory activity is usually limited by several factors, one of which is high level of peroxides present in inflammatory
Inflammation
Inflammation is part of the complex biological response of vascular tissues to harmful stimuli, such as pathogens, damaged cells, or irritants. Inflammation is a protective attempt by the organism to remove the injurious stimuli and to initiate the healing process...
lesions. However, in some circumstances, even peripheral anti-inflammatory activity comparable to other NSAIDs can be observed. An article in nature communications from a research group in Lund, Sweden in November 2011 has found a hint to the analgesic mechanism of acetaminophen (paracetamol), being that the metabolites of acetaminophen e.g NAPQI, act on TRPA1-receptors in the spinal cord to suppress the signal transduction from the superficial layers of the dorsal horn, to alleviate pain.
Because of its selectivity for COX-2 it does not significantly inhibit the production of the pro-clotting thromboxane
Thromboxane
Thromboxane is a member of the family of lipids known as eicosanoids. The two major thromboxanes are thromboxane A2 and thromboxane B2. The distinguishing feature of thromboxanes is a 6-membered ether-containing ring....
s.
The COX family of enzymes are responsible for the metabolism of arachidonic acid
Arachidonic acid
Arachidonic acid is a polyunsaturated omega-6 fatty acid 20:4.It is the counterpart to the saturated arachidic acid found in peanut oil, Arachidonic acid (AA, sometimes ARA) is a polyunsaturated omega-6 fatty acid 20:4(ω-6).It is the counterpart to the saturated arachidic acid found in peanut oil,...
to prostaglandin H2
Prostaglandin H2
Prostaglandin H2 is a type of Prostaglandin which is derived from arachidonic acid and is a precursor for many other biologically significant molecules.It is acted upon by:* prostacyclin synthase to create prostacyclin...
, an unstable molecule that is, in turn, converted to numerous other pro-inflammatory compounds. Classical anti-inflammatories such as the NSAIDs block this step. Only when appropriately oxidized is the COX enzyme highly active.
Paracetamol reduces the oxidized form of the COX enzyme, preventing it from forming pro-inflammatory chemicals. This leads to a reduced amount of Prostaglandin E2 in the CNS, thus lowering the hypothalamic set-point in the thermoregulatory centre.
Paracetamol also modulates the endogenous cannabinoid system
Endocannabinoid system
The endocannabinoid system refers to a group of neuromodulatory lipids and their receptors that are involved in a variety of physiological processes including appetite, pain-sensation, mood, and memory; it mediates the psychoactive effects of cannabis and, broadly speaking, includes:* The...
. Paracetamol is metabolized to AM404
AM404
AM404, also known as N-arachidonoylethanolamide, is an active metabolite of paracetamol , responsible for all or part of its analgesic action.-Pharmacology:...
, a compound with several actions; what is most important is that it inhibits the uptake of the endogenous cannabinoid/vanilloid anandamide
Anandamide
Anandamide, also known as N-arachidonoylethanolamide or AEA, is an endogenous cannabinoid neurotransmitter. The name is taken from the Sanskrit word ananda, which means "bliss, delight", and amide. It is synthesized from N-arachidonoyl phosphatidylethanolamine by multiple pathways...
by neurons. Anandamide uptake would result in the activation of the main pain receptor (nociceptor) of the body, the TRPV1
TRPV1
The transient receptor potential cation channel subfamily V member 1 ', also known as the capsaicin receptor and the vanilloid receptor 1, is a protein that, in humans, is encoded by the TRPV1 gene...
(older name: vanilloid receptor). Furthermore, AM404 inhibits sodium channels, as do the anesthetics lidocaine and procaine. Either of these actions by themselves has been shown to reduce pain, and are a possible mechanism for paracetamol. However, it has been demonstrated that, after blocking cannabinoid receptors with synthetic antagonists, paracetamol's analgesic effects are prevented, suggesting its pain-relieving action involves the endogenous cannabinoid system. Spinal TRPA1
TRPA1
Transient receptor potential cation channel, subfamily A, member 1, also known as TRPA1, is a protein which in humans is encoded by the TRPA1 gene....
receptors have also been demonstrated to mediate antinociceptive effects of paracetamol and Δ9-tetrahydrocannabiorcol in mice.
The exact mechanisms by which COX is inhibited in various circumstances is still a subject of discussion. Because of differences in the activity of paracetamol, aspirin, and other NSAIDs, it has been postulated that further COX variants may exist. A recently discovered COX-1 splice variant termed COX-3
COX-3
COX-3 is an enzyme that is encoded by the PTGS1 gene, but is not functional in humans. COX-3 is the third and most recently discovered cyclooxygenase isozyme, the others being COX-1 and COX-2...
was considered to explain some of the knowledge gap but newer findings do not support the hypothesis that it plays any significant role in the functioning of paracetamol.
Aspirin is known to inhibit the cyclooxygenase
Cyclooxygenase
Cyclooxygenase is an enzyme that is responsible for formation of important biological mediators called prostanoids, including prostaglandins, prostacyclin and thromboxane. Pharmacological inhibition of COX can provide relief from the symptoms of inflammation and pain...
(COX) family of enzymes and, because paracetamol's action is partially similar to aspirin's, much research has focused on whether paracetamol also inhibits COX. It is now clear that paracetamol acts via at least two pathways.
One theory holds that paracetamol works by inhibiting the COX-3
COX-3
COX-3 is an enzyme that is encoded by the PTGS1 gene, but is not functional in humans. COX-3 is the third and most recently discovered cyclooxygenase isozyme, the others being COX-1 and COX-2...
isoform of the COX family of enzymes. When expressed in dogs, this enzyme shares a strong similarity to the other COX enzymes, produces pro-inflammatory chemicals, and is selectively inhibited by paracetamol. However, some research has suggested that, in humans and mice, the COX-3 enzyme is without inflammatory action. Another possibility is that paracetamol blocks cyclooxygenase (as in aspirin), but that is in an inflammatory environment where the concentration of peroxides is high, and the high oxidation state of paracetamol prevents its actions. This would mean that paracetamol has no direct effect at the site of inflammation, but instead acts in the CNS where the environment is not oxidative, to reduce temperature, etc. The exact mechanism by which paracetamol is believed to affect COX-3 is disputed.
Structure and reactivity
Benzene
Benzene is an organic chemical compound. It is composed of 6 carbon atoms in a ring, with 1 hydrogen atom attached to each carbon atom, with the molecular formula C6H6....
ring core, substituted
Substituent
In organic chemistry and biochemistry, a substituent is an atom or group of atoms substituted in place of a hydrogen atom on the parent chain of a hydrocarbon...
by one hydroxyl
Hydroxyl
A hydroxyl is a chemical group containing an oxygen atom covalently bonded with a hydrogen atom. In inorganic chemistry, the hydroxyl group is known as the hydroxide ion, and scientists and reference works generally use these different terms though they refer to the same chemical structure in...
group and the nitrogen
Nitrogen
Nitrogen is a chemical element that has the symbol N, atomic number of 7 and atomic mass 14.00674 u. Elemental nitrogen is a colorless, odorless, tasteless, and mostly inert diatomic gas at standard conditions, constituting 78.08% by volume of Earth's atmosphere...
atom of an amide
Amide
In chemistry, an amide is an organic compound that contains the functional group consisting of a carbonyl group linked to a nitrogen atom . The term refers both to a class of compounds and a functional group within those compounds. The term amide also refers to deprotonated form of ammonia or an...
group in the para (1,4) pattern
Arene substitution patterns
Arene substitution patterns are part of organic chemistry IUPAC nomenclature and pinpoint the position of substituents other than hydrogen in relation to each other on an aromatic hydrocarbon.- Ortho, meta, and para substitution :...
. The amide group is acetamide
Acetamide
Acetamide is an organic compound with the formula CH3CONH2. It is the simplest amide derived from acetic acid. It finds some use as a plasticizer and as an industrial solvent...
(ethanamide). It is an extensively conjugated system
Conjugated system
In chemistry, a conjugated system is a system of connected p-orbitals with delocalized electrons in compounds with alternating single and multiple bonds, which in general may lower the overall energy of the molecule and increase stability. Lone pairs, radicals or carbenium ions may be part of the...
, as the lone pair
Lone pair
In chemistry, a lone pair is a valence electron pair without bonding or sharing with other atoms. They are found in the outermost electron shell of an atom, so lone pairs are a subset of a molecule's valence electrons...
on the hydroxyl oxygen, the benzene pi cloud, the nitrogen lone pair, the p orbital
Atomic orbital
An atomic orbital is a mathematical function that describes the wave-like behavior of either one electron or a pair of electrons in an atom. This function can be used to calculate the probability of finding any electron of an atom in any specific region around the atom's nucleus...
on the carbonyl
Carbonyl
In organic chemistry, a carbonyl group is a functional group composed of a carbon atom double-bonded to an oxygen atom: C=O. It is common to several classes of organic compounds, as part of many larger functional groups....
carbon, and the lone pair on the carbonyl oxygen are all conjugated. The presence of two activating groups also make the benzene ring highly reactive toward electrophilic
Electrophile
In general electrophiles are positively charged species that are attracted to an electron rich centre. In chemistry, an electrophile is a reagent attracted to electrons that participates in a chemical reaction by accepting an electron pair in order to bond to a nucleophile...
aromatic substitution. As the substituents are ortho,para-directing and para with respect to each other, all positions on the ring are more or less equally activated. The conjugation also greatly reduces the basicity
Base (chemistry)
For the term in genetics, see base A base in chemistry is a substance that can accept hydrogen ions or more generally, donate electron pairs. A soluble base is referred to as an alkali if it contains and releases hydroxide ions quantitatively...
of the oxygens and the nitrogen, while making the hydroxyl acidic through delocalisation of charge developed on the phenoxide
Phenol
Phenol, also known as carbolic acid, phenic acid, is an organic compound with the chemical formula C6H5OH. It is a white crystalline solid. The molecule consists of a phenyl , bonded to a hydroxyl group. It is produced on a large scale as a precursor to many materials and useful compounds...
anion
Ion
An ion is an atom or molecule in which the total number of electrons is not equal to the total number of protons, giving it a net positive or negative electrical charge. The name was given by physicist Michael Faraday for the substances that allow a current to pass between electrodes in a...
.
Synthesis
In the laboratory, paracetamol is easily prepared by nitratingNitration
Nitration is a general chemical process for the introduction of a nitro group into a chemical compound. The dominant application of nitration is for the production of nitrobenzene, the precursor to methylene diphenyl diisocyanate...
phenol with sodium nitrate
Sodium nitrate
Sodium nitrate is the chemical compound with the formula NaNO3. This salt, also known as Chile saltpeter or Peru saltpeter to distinguish it from ordinary saltpeter, potassium nitrate, is a white solid which is very soluble in water...
, separating the desired p-nitrophenol
4-Nitrophenol
4-Nitrophenol is a phenolic compound that has a nitro group at the opposite position of hydroxy group on the benzene ring.-Properties:...
from the ortho- byproduct, and reducing the nitro group with sodium borohydride
Sodium borohydride
Sodium borohydride, also known as sodium tetrahydridoborate, is an inorganic compound with the formula NaBH4. This white solid, usually encountered as a powder, is a versatile reducing agent that finds wide application in chemistry, both in the laboratory and on a technical scale. Large amounts are...
. The resultant p-aminophenol is then acetylated with acetic anhydride
Acetic anhydride
Acetic anhydride, or ethanoic anhydride, is the chemical compound with the formula 2O. Commonly abbreviated Ac2O, it is the simplest isolatable acid anhydride and is a widely used reagent in organic synthesis...
. In this reaction, phenol
Phenol
Phenol, also known as carbolic acid, phenic acid, is an organic compound with the chemical formula C6H5OH. It is a white crystalline solid. The molecule consists of a phenyl , bonded to a hydroxyl group. It is produced on a large scale as a precursor to many materials and useful compounds...
is strongly activating, thus the reaction requires only mild conditions (cf. the nitration of benzene). The industrial process is analogous, but hydrogenation is used instead of the sodium borohydride reduction.

A simpler synthesis by Hoechst-Celanese involves direct acylation of phenol with acetic anhydride catalyzed by HF, conversion of the ketone to a ketoxime with hydroxylamine
Hydroxylamine
Hydroxylamine is an inorganic compound with the formula NH2OH. The pure material is a white, unstable crystalline, hygroscopic compound. However, hydroxylamine is almost always provided and used as an aqueous solution. It is used to prepare oximes, an important functional group. It is also an...
, followed by the acid-catalyzed Beckmann rearrangement
Beckmann rearrangement
The Beckmann rearrangement, named after the German chemist Ernst Otto Beckmann , is an acid-catalyzed rearrangement of an oxime to an amide...
to give the amide.
Demand for paracetamol in the United States was estimated at 30–35 thousand tonnes per year in 1997, equal to the demand from the rest of the world.
Metabolism
Drug metabolism
Drug metabolism is the biochemical modification of pharmaceutical substances by living organisms, usually through specialized enzymatic systems. This is a form of xenobiotic metabolism. Drug metabolism often converts lipophilic chemical compounds into more readily excreted polar products...
primarily in the liver
Liver
The liver is a vital organ present in vertebrates and some other animals. It has a wide range of functions, including detoxification, protein synthesis, and production of biochemicals necessary for digestion...
, into non-toxic products. Three metabolic pathway
Metabolic pathway
In biochemistry, metabolic pathways are series of chemical reactions occurring within a cell. In each pathway, a principal chemical is modified by a series of chemical reactions. Enzymes catalyze these reactions, and often require dietary minerals, vitamins, and other cofactors in order to function...
s are notable:
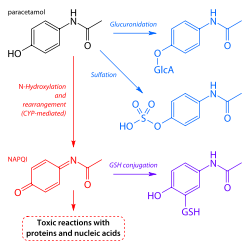
- GlucuronidationGlucuronidationGlucuronidation is the addition of glucuronic acid to a substrate. Glucuronidation is often involved in xenobiotic metabolism of substances such as drugs, pollutants, bilirubin, androgens, estrogens, mineralocorticoids, glucocorticoids, fatty acid derivatives, retinoids, and bile acids...
is believed to account for 40% to two-thirds of the metabolism of paracetamol. - Sulfation (sulfate conjugation) may account for 20–40%.
- N-hydroxylation and rearrangement, then GSH conjugation, accounts for less than 15%. The hepatic cytochrome P450 enzyme system metabolizes paracetamol, forming a minor yet significant alkylating metabolite known as NAPQINAPQINAPQI is a toxic byproduct produced during the xenobiotic metabolism of the analgesic paracetamol...
(N-acetyl-p-benzo-quinone imine). NAPQI is then irreversibly conjugated with the sulfhydryl groupsThiolIn organic chemistry, a thiol is an organosulfur compound that contains a carbon-bonded sulfhydryl group...
of glutathioneGlutathioneGlutathione is a tripeptide that contains an unusual peptide linkage between the amine group of cysteine and the carboxyl group of the glutamate side-chain...
.
All three pathways yield final products that are inactive, non-toxic, and eventually excreted by the kidneys. In the third pathway, however, the intermediate product NAPQI is toxic. NAPQI is primarily responsible for the toxic effects of paracetamol; this constitutes an example of toxication
Toxication
Toxication is the process of metabolism in which the metabolite of a compound is more toxic than the parent drug or chemical. A parent drug or chemical that was previously non-toxic may be called a protoxin.Toxication may involve:...
.
Production of NAPQI is due primarily to two isoenzymes of cytochrome P450: CYP2E1
CYP2E1
Cytochrome P450 2E1 , a member of the cytochrome P450 mixed-function oxidase system, is involved in the metabolism of xenobiotics in the body. In humans, the CYP2E1 enzyme is encoded by the CYP2E1 gene...
and CYP1A2
CYP1A2
Cytochrome P450 1A2 , a member of the cytochrome P450 mixed-function oxidase system, is involved in the metabolism of xenobiotics in the body...
. The P450 gene is highly polymorphic
Polymorphism (biology)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species — in other words, the occurrence of more than one form or morph...
, however, and individual differences in paracetamol toxicity are believed due to a third isoenzyme, CYP2D6
CYP2D6
Cytochrome P450 2D6 , a member of the cytochrome P450 mixed-function oxidase system, is one of the most important enzymes involved in the metabolism of xenobiotics in the body. Also, many substances are bioactivated by CYP2D6 to form their active compounds...
. Genetic polymorphisms in CYP2D6
CYP2D6
Cytochrome P450 2D6 , a member of the cytochrome P450 mixed-function oxidase system, is one of the most important enzymes involved in the metabolism of xenobiotics in the body. Also, many substances are bioactivated by CYP2D6 to form their active compounds...
may contribute to significantly different rates of production of NAPQI. Furthermore, individuals can be classified as "extensive", "ultrarapid", and "poor" metabolizers (producers of NAPQI), depending on their levels of CYP2D6 expression. Although CYP2D6 metabolises paracetamol into NAPQI to a lesser extent than other P450 enzymes, its activity may contribute to paracetamol toxicity in extensive and ultrarapid metabolisers, and when paracetamol is taken at very large doses. At usual doses, NAPQI is quickly detoxified by conjugation. Following overdose, and possibly also in extensive and ultrarapid metabolizers, this detoxification pathway becomes saturated, and, as a consequence, NAPQI accumulates.
Reactions
p-Aminophenol may be obtained by the amide hydrolysisHydrolysis
Hydrolysis is a chemical reaction during which molecules of water are split into hydrogen cations and hydroxide anions in the process of a chemical mechanism. It is the type of reaction that is used to break down certain polymers, especially those made by condensation polymerization...
of paracetamol. p-Aminophenol prepared this way, and related to the commercially available Metol
Metol
Metol is the chemical compound with the name monomethyl-p-aminophenol hemisulfate. It is a developing agent used in black & white photographic developers...
, has been used as a developer in photography by hobbyists. This reaction is also used to determine paracetamol in urine samples: After hydrolysis with hydrochloric acid, p-aminophenol reacts in ammonia solution with a phenol derivate, e.g. salicylic acid, to form an indophenol
Indophenol
Indophenol is an artificial blue metachromatic dye, obtained by the action of phenol on certain nitrogenous derivatives of quinone. Indophenol resembles the color indigo in appearance...
dye under oxidization by air.
History
Cyanosis
Cyanosis is the appearance of a blue or purple coloration of the skin or mucous membranes due to the tissues near the skin surface being low on oxygen. The onset of cyanosis is 2.5 g/dL of deoxyhemoglobin. The bluish color is more readily apparent in those with high hemoglobin counts than it is...
due to methemoglobinemia
Methemoglobinemia
Methemoglobinemia is a disorder characterized by the presence of a higher than normal level of methemoglobin in the blood. Methemoglobin is an oxidized form of hemoglobin that has an increased affinity for oxygen, resulting in a reduced ability to release oxygen to tissues. The oxygen–hemoglobin...
, prompted the search for less toxic aniline derivatives. Harmon Northrop Morse
Harmon Northrop Morse
Harmon Northrop Morse was an American chemist. Today he is known as the first to have synthesized paracetamol, but this substance only became widely used as a drug decades after Morse's death. In the first half of the 20th century he was best known for his study of osmotic pressure, for which he...
had already synthesized paracetamol at Johns Hopkins University
Johns Hopkins University
The Johns Hopkins University, commonly referred to as Johns Hopkins, JHU, or simply Hopkins, is a private research university based in Baltimore, Maryland, United States...
via the reduction of p-nitrophenol
4-Nitrophenol
4-Nitrophenol is a phenolic compound that has a nitro group at the opposite position of hydroxy group on the benzene ring.-Properties:...
with tin
Tin
Tin is a chemical element with the symbol Sn and atomic number 50. It is a main group metal in group 14 of the periodic table. Tin shows chemical similarity to both neighboring group 14 elements, germanium and lead and has two possible oxidation states, +2 and the slightly more stable +4...
in glacial acetic acid
Acetic acid
Acetic acid is an organic compound with the chemical formula CH3CO2H . It is a colourless liquid that when undiluted is also called glacial acetic acid. Acetic acid is the main component of vinegar , and has a distinctive sour taste and pungent smell...
in 1877,
but it was not until 1887 that clinical pharmacologist Joseph von Mering
Joseph von Mering
Josef, Baron von Mering was a German physician.Working at the University of Strasbourg, Mering was the first person to discover that one of the pancreatic functions is the production of insulin, a hormone which controls blood sugar levels.Mering was curious about the...
tried paracetamol on patients. In 1893, von Mering published a paper reporting on the clinical results of paracetamol with phenacetin
Phenacetin
Phenacetin is an analgesic, once widely used; its use has declined because of its adverse effects.-History:Phenacetin was introduced in 1887, and was used principally as an analgesic, and was one of the first synthetic fever reducers to go on the market...
, another aniline derivative. Von Mering claimed that, unlike phenacetin, paracetamol had a slight tendency to produce methemoglobinemia. Paracetamol was then quickly discarded in favor of phenacetin. The sales of phenacetin established Bayer
Bayer
Bayer AG is a chemical and pharmaceutical company founded in Barmen , Germany in 1863. It is headquartered in Leverkusen, North Rhine-Westphalia, Germany and well known for its original brand of aspirin.-History:...
as a leading pharmaceutical company. Overshadowed in part by aspirin
Aspirin
Aspirin , also known as acetylsalicylic acid , is a salicylate drug, often used as an analgesic to relieve minor aches and pains, as an antipyretic to reduce fever, and as an anti-inflammatory medication. It was discovered by Arthur Eichengrun, a chemist with the German company Bayer...
, introduced into medicine by Heinrich Dreser
Heinrich Dreser
Heinrich Dreser was a German chemist, responsible for the aspirin and heroin projects at Bayer AG. He was a key factor in the creation of the widely used modern drug, Codeine. On December 21, 1924, Dreser died of a stroke....
in 1899, phenacetin was popular for many decades, particularly in widely advertised over-the-counter "headache mixtures", usually containing phenacetin, an aminopyrine derivative of aspirin, caffeine, and sometimes a barbiturate
Barbiturate
Barbiturates are drugs that act as central nervous system depressants, and can therefore produce a wide spectrum of effects, from mild sedation to total anesthesia. They are also effective as anxiolytics, as hypnotics, and as anticonvulsants...
.
Von Mering's claims remained essentially unchallenged for half a century, until two teams of researchers from the United States analyzed the metabolism of acetanilide and paracetamol. In 1947 David Lester
David Lester (biochemist)
David Lester was an American biochemist who did extensive studies of alcoholism, and was a professor at Rutgers University.- Life and career :...
and Leon Greenberg found strong evidence that paracetamol was a major metabolite of acetanilide in human blood, and in a subsequent study they reported that large doses of paracetamol given to albino rats did not cause methemoglobinemia. In three papers published in the September 1948 issue of the Journal of Pharmacology and Experimental Therapeutics
Journal of Pharmacology and Experimental Therapeutics
The Journal of Pharmacology and Experimental Therapeutics is a peer-reviewed pharmacology journal published since 1909 by the American Society for Pharmacology and Experimental Therapeutics...
, Bernard Brodie
Bernard Brodie (biochemist)
Bernard Beryl Brodie , a leading researcher on drug therapy, is considered by many to be the founder of modern pharmacology and brought the field to prominence in the 1940s and 1950s. He was a major figure in the field of drug metabolism, the study of how drugs interact in the body and how they...
, Julius Axelrod
Julius Axelrod
Julius Axelrod was an American biochemist. He won a share of the Nobel Prize in Physiology or Medicine in 1970 along with Bernard Katz and Ulf von Euler...
and Frederick Flinn confirmed using more specific methods that paracetamol was the major metabolite of acetanilide in human blood, and established it was just as efficacious an analgesic as its precursor. They also suggested that methemoglobinemia is produced in humans mainly by another metabolite, phenylhydroxylamine
Phenylhydroxylamine
Phenylhydroxylamine is the organic compound with the formula C6H5NHOH. It is an intermediate in the redox-related pair C6H5NH2 and C6H5NO. Phenylhydroxylamine should not be confused with its isomer α-phenylhydroxylamine or O-phenylhydroxylamine, is C6H5ONH2.-Preparation and derivatives:This...
. A followup paper by Brodie and Axelrod in 1949 established that phenacetin was also metabolized to paracetamol. This led to a "rediscovery" of paracetamol. It has been suggested that contamination of paracetamol with 4-aminophenol, the substance von Mering synthesized it from, may be the cause for his spurious findings.
Paracetamol was first marketed in the United States in 1953 by Sterling-Winthrop Co., which promoted it as preferable to aspirin since it was safe to take for children and people with ulcers. The best known brand today for paracetamol in the United States, Tylenol
Tylenol
Tylenol is a North American brand of drugs advertised for reducing pain, reducing fever, and relieving the symptoms of allergies, cold, cough, and flu. The active ingredient of its original, flagship product, paracetamol , is marketed as an analgesic and antipyretic...
, was established in 1955 when McNeil Laboratories
McNeil Laboratories
McNeil Consumer Healthcare is a medicals products company belonging to the Johnson & Johnson healthcare products group.-History:The company was founded on March 16, 1879 by 23-year-old Robert McNeil, who paid $167 for a drugstore complete with fixtures, inventory and soda fountain, as a retail...
started selling paracetamol as a pain and fever reliever for children, under the brand name Tylenol Children's Elixir—the word "tylenol" was a contraction of para-acetylaminophenol. In 1956, 500 mg tablets of paracetamol went on sale in the United Kingdom under the trade name Panadol, produced by Frederick Stearns & Co, a subsidiary of Sterling Drug
Sterling Drug
Sterling Drug was a global pharmaceutical company based in the United States, known as Sterling-Winthrop, Inc. after the merger with Winthrop-Stearns Inc. and then as Sterling Winthrop Pharmaceuticals, whose primary product lines included diagnostic imaging...
Inc. Panadol was originally available only by prescription, for the relief of pain and fever, and was advertised as being "gentle to the stomach," since other analgesic agents of the time contained aspirin, a known stomach irritant. In 1963, paracetamol was added to the British Pharmacopoeia
British Pharmacopoeia
The British Pharmacopoeia is an annual published collection of quality standards for UK medicinal substances. It is used by individuals and organizations involved in pharmaceutical research, development, manufacture and testing....
, and has gained popularity since then as an analgesic agent with few side-effects and little interaction with other pharmaceutical agents. Concerns about paracetamol's safety delayed its widespread acceptance until the 1970s, but in the 1980s paracetamol sales exceeded those of aspirin in many countries, including the United Kingdom. This was accompanied by the commercial demise of phenacetin, blamed as the cause of analgesic nephropathy and hematological toxicity.
The U.S. patent
Patent
A patent is a form of intellectual property. It consists of a set of exclusive rights granted by a sovereign state to an inventor or their assignee for a limited period of time in exchange for the public disclosure of an invention....
on paracetamol has long expired, and generic versions of the drug are widely available under the Drug Price Competition and Patent Term Restoration Act
Drug Price Competition and Patent Term Restoration Act
-Overview:The Drug Price Competition and Patent Term Restoration Act, informally known as the "Hatch-Waxman Act" [Public Law 98-417], is a 1984 United States federal law which established the modern system of generic drugs...
of 1984, although certain Tylenol preparations were protected until 2007. U.S. patent 6,126,967 filed September 3, 1998 was granted for "Extended release acetaminophen particles".
Available forms
Paracetamol is available in a tabletTablet
A tablet is a pharmaceutical dosage form. It comprises a mixture of active substances and excipients, usually in powder form, pressed or compacted from a powder into a solid dose...
, capsule, liquid suspension, suppository
Suppository
A suppository is a drug delivery system that is inserted into the rectum , vagina or urethra , where it dissolves.They are used to deliver both systemically-acting and locally-acting medications....
, intravenous, and intramuscular form. The common adult dose is 500 mg to 1000 mg. The recommended maximum daily dose, for adults, is 4000 mg. In recommended doses, paracetamol generally is safe for children and infants, as well as for adults, although rare cases of acute liver injury have been linked to amounts lower than 2500 mg per day.
Panadol
Panadol
Panadol is the GlaxoSmithKline trade name for paracetamol or acetaminophen , which is an over the counter pharmaceutical analgesic and antipyretic ....
, which is marketed in Africa, Asia, Europe, Central America, and Australasia
Australasia
Australasia is a region of Oceania comprising Australia, New Zealand, the island of New Guinea, and neighbouring islands in the Pacific Ocean. The term was coined by Charles de Brosses in Histoire des navigations aux terres australes...
, is the most widely available brand of paracetamol, sold in over 80 countries. In North America, paracetamol is sold in generic form (usually labeled as acetaminophen) or under a number of trade names, for instance, Tylenol
Tylenol
Tylenol is a North American brand of drugs advertised for reducing pain, reducing fever, and relieving the symptoms of allergies, cold, cough, and flu. The active ingredient of its original, flagship product, paracetamol , is marketed as an analgesic and antipyretic...
(McNeil-PPC, Inc.
McNeil Laboratories
McNeil Consumer Healthcare is a medicals products company belonging to the Johnson & Johnson healthcare products group.-History:The company was founded on March 16, 1879 by 23-year-old Robert McNeil, who paid $167 for a drugstore complete with fixtures, inventory and soda fountain, as a retail...
), Anacin-3
Anacin
Anacin is a pain reliever intended for the temporary relief of minor aches and pains. Anacin is a product of Insight Pharmaceuticals. Anacin's active ingredients are aspirin and caffeine.-History:...
, Tempra, Datril, and Ofirmev. While there is brand named paracetamol available in the UK (e.g. Panadol), unbranded or generic paracetamol is more commonly sold. Acamol
Acamol
Acamol is a brand name for paracetamol. Acamol is produced by Teva Pharmaceutical Industries. Each Acamol tablet contains 500 mg of paracetamol. Acamol is the most popular drug in Israel....
, a brand name for paracetamol produced by Teva Pharmaceutical Industries
Teva Pharmaceutical Industries
Teva Pharmaceutical Industries Ltd. , is an international pharmaceutical company headquartered in Petah Tikva, Israel. It specializes in generic and proprietary pharmaceuticals and active pharmaceutical ingredients...
in Israel
Israel
The State of Israel is a parliamentary republic located in the Middle East, along the eastern shore of the Mediterranean Sea...
, is one of the most widely used drugs in that country. In the Philippines, the largest-selling paracetamol brand is Biogesic, manufactured by the drug giant United Laboratories. Biogesic tablet sales reach nearly a billion units each year in the country alone, not including liquid suspension formats. The brand is also available in most of the ASEAN countries where the drug giant has market presence.
In Europe, the most common brands of paracetamol are Efferalgan and Doliprane. In India, the most common brand of paracetamol is Crocin manufactured by Glaxo SmithKline Asia. In Bangladesh the most popular brand is Napa manufactured by Beximco Pharma. In China paracetamol is sold over the counter as Duìyǐxiān'ānjīfēn Piàn (对乙酰氨基酚片). Likewise in Japan it is sold under the name Acetaminophen (アセトアミノフェン Asetoaminofen). In North Korea the DPRK-Swiss joint venture PyongSu Pharma
Pyongsu Joint Venture Company
Pyongsu Joint Venture Company, Ltd is a Pharmaceutical company jointly founded in Pyongyang, DPRK .-Founders & Shareholders:*Pyongyang Pharmaceutical Company*Ministry of Public Health...
markets the drug as PyongSu Cetamol.
In some formulations, paracetamol is combined with the opioid
Opioid
An opioid is a psychoactive chemical that works by binding to opioid receptors, which are found principally in the central and peripheral nervous system and the gastrointestinal tract...
codeine
Codeine
Codeine or 3-methylmorphine is an opiate used for its analgesic, antitussive, and antidiarrheal properties...
, sometimes referred to as co-codamol
Co-codamol
Co-codamol is a non-proprietary name used to denote a compound analgesic, a combination of codeine phosphate and paracetamol . Co-codamol Co-codamol (BAN) is a non-proprietary name used to denote a compound analgesic, a combination of codeine phosphate and paracetamol (acetaminophen). Co-codamol...
(BAN
British Approved Name
A British Approved Name is the official non-proprietary or generic name given to a pharmaceutical substance, as defined in the British Pharmacopoeia...
). In the United States and Canada, this is marketed under the name of Tylenol #1/2/3/4, which contain 8–10 mg, 15 mg, 30 mg, and 60 mg of codeine
Codeine
Codeine or 3-methylmorphine is an opiate used for its analgesic, antitussive, and antidiarrheal properties...
, respectively. In the U.S., this combination is available only by prescription, while the lowest-strength preparation is over-the-counter in Canada, and, in other countries, other strengths may be available over the counter. There are generic forms of these combinations as well. In the UK and in many other countries, this combination is marketed under the names of Tylex CD and Panadeine. Other names include Captin, Disprol, Dymadon, Fensum, Hedex, Mexalen, Nofedol, Paralen, Pediapirin, Perfalgan, and Solpadeine. Paracetamol is also combined with other opioids such as dihydrocodeine
Dihydrocodeine
Dihydrocodeine, also called DHC, Drocode, Paracodeine and Parzone and known by the brand names of Synalgos DC, Panlor DC, Panlor SS, Contugesic, New Bron Solution-ACE, Huscode, Drocode, Paracodin, Codidol, Didor Continus, Dicogesic, Codhydrine, Dekacodin, DH-Codeine,...
, referred to as co-dydramol
Co-dydramol
Co-dydramol is a non-proprietary name used to denote a compound analgesic, a combination of dihydrocodeine tartrate and paracetamol. Co-dydramol tablets are used for the relief of moderate pain...
(BAN
British Approved Name
A British Approved Name is the official non-proprietary or generic name given to a pharmaceutical substance, as defined in the British Pharmacopoeia...
), oxycodone
Oxycodone
Oxycodone is an opioid analgesic medication synthesized from opium-derived thebaine. It was developed in 1916 in Germany, as one of several new semi-synthetic opioids in an attempt to improve on the existing opioids: morphine, diacetylmorphine , and codeine.Oxycodone oral medications are generally...
or hydrocodone
Hydrocodone
Hydrocodone or dihydrocodeinone is a semi-synthetic opioid derived from either of two naturally occurring opiates: codeine and thebaine. It is an orally active narcotic analgesic and antitussive...
, marketed in the U.S. as Percocet
Percocet
The combination oxycodone/paracetamol is a narcotic pain reliever used to treat moderate to severe acute pain, marketed by Endo Pharmaceuticals.-History:The U.S...
and Vicodin
Vicodin
Hydrocodone/paracetamol is a combination of two analgesic products hydrocodone and paracetamol used to relieve moderate to severe pain...
, respectively. Another very commonly used analgesic combination includes paracetamol in combination with propoxyphene napsylate, sold under the brand name Darvocet. A combination of paracetamol, codeine, and the calmative doxylamine succinate
Doxylamine
Doxylamine is one of the many sedating antihistamines used by itself as a short-term sedative, and in combination with other drugs as a night-time cold and allergy relief drug...
is marketed as Syndol or Mersyndol. The efficacy of paracetamol/codeine combinations have been questioned by recent research.
Paracetamol is commonly used in multi-ingredient preparations for migraine
Migraine
Migraine is a chronic neurological disorder characterized by moderate to severe headaches, and nausea...
headache, typically including butalbital
Butalbital
Butalbital, structure presents as 5-allyl-5-isobutylbarbituric acid, is a barbiturate with an intermediate duration of action. It has the same chemical formula as talbutal but a different structure. Butalbital is often combined with other medications, such as paracetamol or aspirin, and is...
and paracetamol with or without caffeine
Caffeine
Caffeine is a bitter, white crystalline xanthine alkaloid that acts as a stimulant drug. Caffeine is found in varying quantities in the seeds, leaves, and fruit of some plants, where it acts as a natural pesticide that paralyzes and kills certain insects feeding on the plants...
, and sometimes containing codeine.
Veterinary use
Paracetamol is extremely toxic to cats. Cats lack the necessary glucuronyl transferase enzymes to safely break paracetamol down, and minute portions of a tablet may prove fatal. Initial symptoms include vomiting, salivation, and discolouration of the tongue and gums. Unlike an overdose in humans, liver damage is rarely the cause of death; instead, methemoglobin formation and the production of Heinz bodies in red blood cells inhibit oxygen transport by the blood, causing asphyxiation (methemoglobemia and hemolytic anemiaHemolytic anemia
Hemolytic anemia is a form of anemia due to hemolysis, the abnormal breakdown of red blood cells , either in the blood vessels or elsewhere in the human body . It has numerous possible causes, ranging from relatively harmless to life-threatening...
). Treatment with N-acetylcysteine, methylene blue
Methylene blue
Methylene blue is a heterocyclic aromatic chemical compound with the molecular formula C16H18N3SCl. It has many uses in a range of different fields, such as biology and chemistry. At room temperature it appears as a solid, odorless, dark green powder, that yields a blue solution when dissolved in...
or both is sometimes effective after the ingestion of small doses of paracetamol.
Although paracetamol is believed to have no significant anti-inflammatory activity, it has been reported as effective as aspirin in the treatment of musculoskeletal pain in dogs. A paracetamol-codeine product (trade name Pardale-V) licensed for use in dogs is available on veterinary prescription in the UK. It should be administered to dogs only on veterinary advice and with extreme caution. The main effects of toxicity in dogs is liver damage, GI ulceration has been reported. N-acetylcysteine treatment is efficacious in dogs when administered within a few hours of paracetamol ingestion.
Paracetamol is also lethal to snakes, and has been suggested as a chemical control program for the invasive brown tree snake
Brown tree snake
The brown tree snake is an arboreal rear-fanged colubrid snake native to eastern and northern coastal Australia, Papua New Guinea, and a large number of islands in northwestern Melanesia....
(Boiga irregularis) in Guam
Guam
Guam is an organized, unincorporated territory of the United States located in the western Pacific Ocean. It is one of five U.S. territories with an established civilian government. Guam is listed as one of 16 Non-Self-Governing Territories by the Special Committee on Decolonization of the United...
.
External links
- Paracetamol at Chemsynthesis
- Paracetamol Information Centre
- The Julius Axelrod Papers
- FDA: Safe Use of Over-the-Counter Pain Relievers/Fever Reducers
- FDA: Consumer Update "Acetaminophen and Liver Injury: Q and A for Consumers" (link)
- FDA: Consumer Update "Acetaminophen and Liver Injury: Q and A for Consumers" (PDF)
- U.S. National Library of Medicine: Drug Information Portal–Paracetamol