
NF-kB
Encyclopedia
Transcription factor
In molecular biology and genetics, a transcription factor is a protein that binds to specific DNA sequences, thereby controlling the flow of genetic information from DNA to mRNA...
of DNA
DNA
Deoxyribonucleic acid is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms . The DNA segments that carry this genetic information are called genes, but other DNA sequences have structural purposes, or are involved in...
. NF-κB is found in almost all animal cell types and is involved in cellular responses to stimuli such as stress, cytokine
Cytokine
Cytokines are small cell-signaling protein molecules that are secreted by the glial cells of the nervous system and by numerous cells of the immune system and are a category of signaling molecules used extensively in intercellular communication...
s, free radicals, ultraviolet irradiation, oxidized LDL, and bacterial or viral antigen
Antigen
An antigen is a foreign molecule that, when introduced into the body, triggers the production of an antibody by the immune system. The immune system will then kill or neutralize the antigen that is recognized as a foreign and potentially harmful invader. These invaders can be molecules such as...
s. NF-κB plays a key role in regulating the immune response to infection (kappa light chains
Immunoglobulin light chain
]The immunoglobulin light chain is the small polypeptide subunit of an antibody .A typical antibody is composed of two immunoglobulin heavy chains and two Ig light chains.-In humans:...
are critical components of immunoglobulins). Incorrect regulation of NF-κB has been linked to cancer, inflammatory and autoimmune diseases, septic shock
Septic shock
Septic shock is a medical emergency caused by decreased tissue perfusion and oxygen delivery as a result of severe infection and sepsis, though the microbe may be systemic or localized to a particular site. It can cause multiple organ dysfunction syndrome and death...
, viral infection, and improper immune development. NF-κB has also been implicated in processes of synaptic plasticity and memory.
Discovery
NF-κB was first discovered in the lab of Nobel PrizeNobel Prize
The Nobel Prizes are annual international awards bestowed by Scandinavian committees in recognition of cultural and scientific advances. The will of the Swedish chemist Alfred Nobel, the inventor of dynamite, established the prizes in 1895...
laureate David Baltimore
David Baltimore
David Baltimore is an American biologist, university administrator, and Nobel laureate in Physiology or Medicine. He served as president of the California Institute of Technology from 1997 to 2006, and is currently the Robert A. Millikan Professor of Biology at Caltech...
via its interaction with an 11-base pair sequence in the immunoglobulin light-chain enhancer
Enhancer (genetics)
In genetics, an enhancer is a short region of DNA that can be bound with proteins to enhance transcription levels of genes in a gene cluster...
in B cells.
Structure
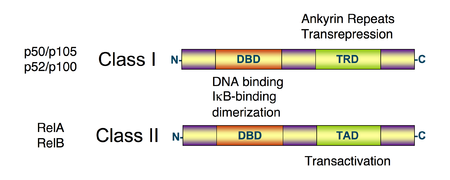
All proteins of the NF-κB family share a Rel homology domainRel homology domain
The Rel homology domain is a protein domain found in a family of eukaryotic transcription factors, which includes NF-κB, NFAT, among others. Some of these transcription factors appear to form multi-protein DNA-bound complexes...
in their N-terminus. A subfamily of NF-κB proteins, including RelA, RelB, and c-Rel, have a transactivation
Transactivation
In molecular biology and genetics, transactivation is an increased rate of gene expression triggered either by biological processes or by artificial means.- Natural transactivation :...
domain in their C-termini. In contrast, the NF-κB1 and NF-κB2 proteins are synthesized as large precursors, p105, and p100, which undergo processing to generate the mature NF-κB subunits, p50 and p52, respectively. The processing of p105 and p100 is mediated by the ubiquitin
Ubiquitin
Ubiquitin is a small regulatory protein that has been found in almost all tissues of eukaryotic organisms. Among other functions, it directs protein recycling.Ubiquitin can be attached to proteins and label them for destruction...
/proteasome
Proteasome
Proteasomes are very large protein complexes inside all eukaryotes and archaea, and in some bacteria. In eukaryotes, they are located in the nucleus and the cytoplasm. The main function of the proteasome is to degrade unneeded or damaged proteins by proteolysis, a chemical reaction that breaks...
pathway and involves selective degradation of their C-terminal region containing ankyrin repeats. Whereas the generation of p52 from p100 is a tightly-regulated process, p50 is produced from constitutive processing of p105.
Members
NF-κB family members share structural homologyHomology (biology)
Homology forms the basis of organization for comparative biology. In 1843, Richard Owen defined homology as "the same organ in different animals under every variety of form and function". Organs as different as a bat's wing, a seal's flipper, a cat's paw and a human hand have a common underlying...
with the retroviral oncoprotein
Oncogene
An oncogene is a gene that has the potential to cause cancer. In tumor cells, they are often mutated or expressed at high levels.An oncogene is a gene found in the chromosomes of tumor cells whose activation is associated with the initial and continuing conversion of normal cells into cancer...
v-Rel, resulting in their classification as NF-κB/Rel proteins.
There are five proteins in the mammalian NF-κB family:
| Class | Protein | Aliases | Gene |
|---|---|---|---|
| I | NF-κB1 | p105 → p50 | NFKB1 NFKB1 Nuclear factor NF-kappa-B p105 subunit is a protein that in humans is encoded by the NFKB1 gene.This gene encodes a 105 kD protein which can undergo cotranslational processing by the 26S proteasome to produce a 50 kD protein. The 105 kD protein is a Rel protein-specific transcription inhibitor and... |
| NF-κB2 | p100 → p52 | NFKB2 NFKB2 Nuclear factor NF-kappa-B p100 subunit is a protein that in humans is encoded by the NFKB2 gene.-Interactions:NFKB2 has been shown to interact with NFKBIE, BCL3, MAP3K8, BTRC, RELA, RELB, NFKB1, REL and TSC22D3.-Further reading:... |
|
| II | RelA | p65 | RELA RELA Transcription factor p65 is a protein that in humans is encoded by the RELA gene.-Interactions:RELA has been shown to interact with NFKBIB, ETHE1, NFKBIE, RFC1, TRIB3, CREB binding protein, Neutrophil cytosolic factor 1, Glucocorticoid receptor, MTPN, BRCA1, C-Fos, POU2F1, BTRC, TATA-binding... |
| RelB | RELB RELB Transcription factor RelB is a protein that in humans is encoded by the RELB gene.-Interactions:RELB has been shown to interact with NFKB2 and NFKB1.-References:... |
||
| c-Rel | REL REL The proto-oncogene c-Rel is a protein that in humans is encoded by the REL gene. The c-Rel protein is a member of the NF-κB family of transcription factors and contains a Rel homology domain at its N-terminus and two C-terminal transactivation domains. c-Rel has an important role in B-cell... |
Below are the five human NF-κB family members:
Species distribution and evolution
In addition to mammals, NF-κB is found in a number of simple animals as well. These include cnidariaCnidaria
Cnidaria is a phylum containing over 9,000 species of animals found exclusively in aquatic and mostly marine environments. Their distinguishing feature is cnidocytes, specialized cells that they use mainly for capturing prey. Their bodies consist of mesoglea, a non-living jelly-like substance,...
ns (such as sea anemone
Sea anemone
Sea anemones are a group of water-dwelling, predatory animals of the order Actiniaria; they are named after the anemone, a terrestrial flower. Sea anemones are classified in the phylum Cnidaria, class Anthozoa, subclass Zoantharia. Anthozoa often have large polyps that allow for digestion of larger...
s, coral
Coral
Corals are marine animals in class Anthozoa of phylum Cnidaria typically living in compact colonies of many identical individual "polyps". The group includes the important reef builders that inhabit tropical oceans and secrete calcium carbonate to form a hard skeleton.A coral "head" is a colony of...
and hydra
Hydra
Hydra is the name of the Lernaean Hydra, a many-headed serpent in Greek mythology."Hydra" may also refer to:- Astronomy :* Hydra , the largest of the modern star constellations* Hydra , a satellite of Pluto...
), porifera (sponges), the single-celled eukaryote Capsaspora owczarzaki and insect
Insect
Insects are a class of living creatures within the arthropods that have a chitinous exoskeleton, a three-part body , three pairs of jointed legs, compound eyes, and two antennae...
s (such as moth
Moth
A moth is an insect closely related to the butterfly, both being of the order Lepidoptera. Moths form the majority of this order; there are thought to be 150,000 to 250,000 different species of moth , with thousands of species yet to be described...
s, mosquito
Mosquito
Mosquitoes are members of a family of nematocerid flies: the Culicidae . The word Mosquito is from the Spanish and Portuguese for little fly...
es and fruitflies). The sequencing of the genomes of the mosquitoes A. aegypti
Aedes aegypti
The yellow fever mosquito, Aedes aegypti is a mosquito that can spread the dengue fever, Chikungunya and yellow fever viruses, and other diseases. The mosquito can be recognized by white markings on legs and a marking in the form of a lyre on the thorax...
and A. gambiae
Anopheles gambiae
Anopheles gambiae is a complex of at least seven morphologically distinguishable species of mosquitoes in the genus Anopheles. This complex was recognised in the 1960s and includes the most important vectors of malaria in sub-Saharan Africa and the most efficient malaria vectors known.This species...
, and the fruitfly D. melanogaster
Drosophila melanogaster
Drosophila melanogaster is a species of Diptera, or the order of flies, in the family Drosophilidae. The species is known generally as the common fruit fly or vinegar fly. Starting from Charles W...
has allowed comparative genetic and evolutionary studies on NF-κB. In those insect species, activation of NF-κB is triggered by the Toll pathway
Toll-like receptor
Toll-like receptors are a class of proteins that play a key role in the innate immune system. They are single, membrane-spanning, non-catalytic receptors that recognize structurally conserved molecules derived from microbes...
(which evolved independently in insects and mammals) and by the Imd (immune deficiency) pathway.
Activation
NF-κB is important in regulating cellular responses because it belongs to the category of "rapid-acting" primary transcription factors, i.e., transcription factors that are present in cells in an inactive state and do not require new protein synthesis to be activated (other members of this family include transcription factors such as c-JunC-jun
c-Jun is the name of a gene and protein that, in combination with c-Fos, forms the AP-1 early response transcription factor. It was first identified as the Fos-binding protein p39 and only later rediscovered as the product of the c-jun gene. It is activated through double phosphorylation by the...
, STAT
STAT
STAT may mean:*STAT protein, the Signal Transducers and Activators of Transcription protein*Special Tertiary Admissions Test, a set of tests aimed at assessing the critical reasoning abilities of university applicants who lack other formal qualifications...
s, and nuclear hormone receptors). This allows NF-κB to be a first responder to harmful cellular stimuli. Known inducers of NF-κB activity are highly variable and include reactive oxygen species (ROS
Reactive oxygen species
Reactive oxygen species are chemically reactive molecules containing oxygen. Examples include oxygen ions and peroxides. Reactive oxygen species are highly reactive due to the presence of unpaired valence shell electrons....
), tumor necrosis factor alpha (TNFα), interleukin 1-beta (IL-1β
IL1B
Interleukin-1 beta also known as catabolin, is a cytokine protein that in humans is encoded by the IL1B gene. IL-1β precursor is cleaved by caspase 1 . Cytosolic thiol protease cleaves the product to form mature IL-1β.- Function :Interleukin 1 was discovered by Gery in 1972...
), bacterial lipopolysaccharides (LPS
Lipopolysaccharide
Lipopolysaccharides , also known as lipoglycans, are large molecules consisting of a lipid and a polysaccharide joined by a covalent bond; they are found in the outer membrane of Gram-negative bacteria, act as endotoxins and elicit strong immune responses in animals.-Functions:LPS is the major...
), isoproterenol, cocaine
Cocaine
Cocaine is a crystalline tropane alkaloid that is obtained from the leaves of the coca plant. The name comes from "coca" in addition to the alkaloid suffix -ine, forming cocaine. It is a stimulant of the central nervous system, an appetite suppressant, and a topical anesthetic...
, and ionizing radiation
Ionizing radiation
Ionizing radiation is radiation composed of particles that individually have sufficient energy to remove an electron from an atom or molecule. This ionization produces free radicals, which are atoms or molecules containing unpaired electrons...
.
Receptor activator of nuclear factor kappa B (RANK
RANK
Receptor Activator of Nuclear Factor κ B , also known as TRANCE Receptor, is a type I membrane protein that is expressed on the surface of osteoclasts and is involved in their activation upon ligand binding...
), which is a type of TNFR
Tumor necrosis factor receptor
A tumor necrosis factor receptor , or death receptor, is a trimeric cytokine receptor that binds tumor necrosis factors . The receptor cooperates with an adaptor protein , which is important in determining the outcome of the response A tumor necrosis factor receptor (TNFR), or death receptor, is a...
, is a central activator of NF-κB. Osteoprotegerin
Osteoprotegerin
Osteoprotegerin , also known as osteoclastogenesis inhibitory factor , or tumor necrosis factor receptor superfamily member 11B , is a protein that in humans is encoded by the TNFRSF11B gene...
(OPG), which is a decoy receptor homolog for RANK ligand, inhibits RANK by binding to RANKL, and, thus, osteoprotegerin is tightly involved in regulating NF-κB activation.
Many bacterial products and stimulation of a wide variety of cell-surface receptor
Receptor (biochemistry)
In biochemistry, a receptor is a molecule found on the surface of a cell, which receives specific chemical signals from neighbouring cells or the wider environment within an organism...
s lead to NF-κB activation and fairly rapid changes in gene expression. The identification of Toll-like receptor
Toll-like receptor
Toll-like receptors are a class of proteins that play a key role in the innate immune system. They are single, membrane-spanning, non-catalytic receptors that recognize structurally conserved molecules derived from microbes...
s (TLRs) as specific pattern recognition molecules and the finding that stimulation of TLRs leads to activation of NF-κB improved our understanding of how different pathogens activate NF-κB. For example, studies have identified TLR4 as the receptor for the LPS component of Gram-Negative bacteria. TLRs are key regulators of both innate and adaptive immune responses.
Unlike RelA, RelB, and c-Rel, the p50 and p52 NF-κB subunits do not contain transactivation domains in their C terminal halves. Nevertheless, the p50 and p52 NF-κB members play critical roles in modulating the specificity of NF-κB function. Although homodimers of p50 and p52 are, in general, repressors of κB site transcription, both p50 and p52 participate in target gene transactivation by forming heterodimers with RelA, RelB, or c-Rel. In addition, p50 and p52 homodimers also bind to the nuclear protein Bcl-3, and such complexes can function as transcriptional activators.
Inhibition
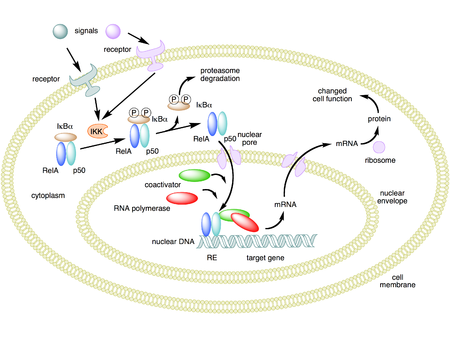
In unstimulated cells, the NF-κB dimers are sequestered in the cytoplasmCytoplasm
The cytoplasm is a small gel-like substance residing between the cell membrane holding all the cell's internal sub-structures , except for the nucleus. All the contents of the cells of prokaryote organisms are contained within the cytoplasm...
by a family of inhibitors, called IκBs (Inhibitor of κB), which are proteins that contain multiple copies of a sequence called ankyrin repeats. By virtue of their ankyrin repeat domains, the IκB proteins mask the nuclear localization signal
Nuclear localization signal
A nuclear localization signal or sequence is an amino acid sequence which 'tags' a protein for import into the cell nucleus by nuclear transport. Typically, this signal consists of one or more short sequences of positively charged lysines or arginines exposed on the protein surface. Different...
s (NLS) of NF-κB proteins and keep them sequestered in an inactive state in the cytoplasm.
IκBs are a family of related proteins that have an N-terminal regulatory domain, followed by six or more ankyrin repeats and a PEST domain
PEST sequence
A PEST sequence is a peptide sequence which is rich in proline , glutamic acid , serine , and threonine . This sequence is associated with proteins that have a short intracellular half-life; hence, it is hypothesized that the PEST sequence acts as a signal peptide for protein degradation.The...
near their C terminus. Although the IκB family consists of IκBα, IκBβ
NFKBIB
NF-kappa-B inhibitor beta is a protein that in humans is encoded by the NFKBIB gene.-Interactions:NFKBIB has been shown to interact with RELA, Retinoid X receptor alpha and IKK2.-Further reading:...
, IκBε
NFKBIE
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, epsilon, also known as NFKBIE, is a protein which in humans is encoded by the NFKBIE gene.- Function :...
, and Bcl-3
BCL3
B-cell lymphoma 3-encoded protein is a protein that in humans is encoded by the BCL3 gene.This gene is a proto-oncogene candidate. It is identified by its translocation into the immunoglobulin alpha-locus in some cases of B-cell leukemia. The protein encoded by this gene contains seven ankyrin...
, the best-studied and major IκB protein is IκBα. Due to the presence of ankyrin repeats in their C-terminal halves, p105 and p100 also function as IκB proteins. The c-terminal half of p100, that is often referred to as IκBδ, also functions as an inhibitor. IκBδ degradation in response to developmental stimuli, such as those transduced through LTβR
Lymphotoxin beta receptor
Lymphotoxin beta receptor is a receptor for lymphotoxin which in humans is encoded by the LTBR gene.-Function:The protein encoded by this gene is a member of the tumor necrosis factor family of receptors. It is expressed on the surface of most cell types, including cells of epithelial and myeloid...
, potentiate NF-κB dimer activation in a NIK dependent non-canonical pathway.
Activation of the NF-κB is initiated by the signal-induced degradation of IκB proteins. This occurs primarily via activation of a kinase called the IκB kinase
IκB kinase
The IκB kinase is an enzyme complex that is involved in propagating the cellular response to inflammation.The IκB kinase enzyme complex is part of the upstream NF-κB signal transduction cascade...
(IKK). IKK is composed of a heterodimer of the catalytic IKK alpha and IKK beta subunits and a "master" regulatory protein termed NEMO
IKBKG
NF-kappa-B essential modulator also known as inhibitor of nuclear factor kappa-B kinase subunit gamma is a protein that in humans is encoded by the IKBKG gene. NEMO is a subunit of the IκB kinase that activates NF-κB. The human gene for IKBKG is located on chromosome Xq28...
(NF-κB essential modulator) or IKK gamma. When activated by signals, usually coming from the outside of the cell, the IκB kinase phosphorylates two serine residues located in an IκB regulatory domain. When phosphorylated on these serines (e.g., serines 32 and 36 in human IκBα), the IκB inhibitor molecules are modified by a process called ubiquitination, which then leads them to be degraded by a cell structure called the proteasome.
With the degradation of IκB, the NF-κB complex is then freed to enter the nucleus where it can 'turn on' the expression of specific genes that have DNA-binding sites for NF-κB nearby. The activation of these genes by NF-κB then leads to the given physiological response, for example, an inflammatory or immune response, a cell survival response, or cellular proliferation. NF-κB turns on expression of its own repressor, IκBα. The newly synthesized IκBα then re-inhibits NF-κB and, thus, forms an auto feedback loop, which results in oscillating levels of NF-κB activity. In addition, several viruses, including the AIDS virus HIV, have binding sites for NF-κB that controls the expression of viral genes, which in turn contribute to viral replication or viral pathogenicity. In the case of HIV-1, activation of NF-κB may, at least in part, be involved in activation of the virus from a latent, inactive state. YopP is a factor secreted by Yersinia
Yersinia
Yersinia is a genus of bacteria in the family Enterobacteriaceae. Yersinia are Gram-negative rod shaped bacteria, a few micrometers long and fractions of a micrometer in diameter, and are facultative anaerobes. Some members of Yersinia are pathogenic in humans; in particular, Y. pestis is the...
pestis, the causative agent of plague, that prevents the ubiquitination of IκB. This causes this pathogen to effectively inhibit the NF-κB pathway and thus block the immune response of a human infected with Yersinia.
Inhibitors of NF-kB activity
Concerning known protein inhibitors of NF-kB activity, one of them is IFRD1IFRD1
Interferon-related developmental regulator 1 is a protein that in humans is encoded by the IFRD1 gene. The gene is expressed mostly in neutrophils, skeletal and cardiac muscle, brain, pancreas....
, which represses the activity of NF-kB p65 by enhancing the HDAC-mediated deacetylation of the p65 subunit at lysine 310, by favoring the recruitment of HDAC3 to p65. In fact IFRD1 forms trimolecular complexes with p65 and HDAC3.
Non-canonical
A select set of cell-differentiating or developmental stimuli, such as lymphotoxin-αLymphotoxin alpha
Lymphotoxin-alpha is a protein that in humans is encoded by the LTA gene.- Function :Lymphotoxin alpha, a member of the tumor necrosis factor family, is a cytokine produced by lymphocytes. LTA is highly inducible, secreted, and exists as homotrimeric molecule. LTA forms heterotrimers with...
, BAFF
B-cell activating factor
B-cell activating factor also known as tumor necrosis factor ligand superfamily member 13B is a protein that in humans is encoded by the TNFLSF13B gene...
or RANKL
RANKL
Receptor activator of nuclear factor kappa-B ligand , also known as tumor necrosis factor ligand superfamily member 11 , TNF-related activation-induced cytokine , osteoprotegerin ligand , and osteoclast differentiation factor , is a protein that in humans is encoded by the TNFSF11 gene.RANKL is...
, activate the non-canonical NF-κB pathway to induce NF-κB/RelB:p52 dimer in the nucleus. In this pathway, activation of the NF-κB inducing kinase (NIK) upon receptor ligation led to the phosphorylation and subsequent proteasomal processing of the NF-κB2 precursor protein p100 into mature p52 subunit in a IKK1/IKKa dependent manner. Then p52 dimerizes with RelB to appear as a nuclear RelB:p52 DNA binding activity and regulate a distinct class of genes. In contrast to the canonical signaling that relies upon NEMO-IKK2 mediated degradation of IκBα, -β, -ε, the non-canonical signaling critically depends on NIK mediated processing of p100 into p52. Given their distinct regulations, these two pathways were thought to be independent of each other. However, recent analyses revealed that synthesis of the constituents of the non-canonical pathway, viz RelB and p52, is controlled by the canonical IKK2-IκB-RelA:p50 signaling. Moreover, generation of the canonical and non-canonical dimers, viz RelA:p50 and RelB:p52, within the cellular milieu are also mechanistically interlinked. These analyses suggest that an integrated NF-κB system network underlies activation of both RelA and RelB containing dimer and that a malfunctioning canonical pathway will lead to an aberrant cellular response also through the non-canonical pathway.
In immunity
NF-κB is a major transcription factor that regulates genes responsible for both the innateInnate immune system
The innate immune system, also known as non-specific immune system and secondary line of defence, comprises the cells and mechanisms that defend the host from infection by other organisms in a non-specific manner...
and adaptive immune response
Adaptive immune system
The adaptive immune system is composed of highly specialized, systemic cells and processes that eliminate or prevent pathogenic growth. Thought to have arisen in the first jawed vertebrates, the adaptive or "specific" immune system is activated by the “non-specific” and evolutionarily older innate...
. Upon activation of either the T- or B-cell receptor, NF-κB becomes activated through distinct signaling components. Upon ligation of the T-cell receptor, protein kinase Lck
Lck
Lck is a protein that is found inside specialized cells of the immune system called lymphocytes. Lck is a tyrosine kinase, which phosphorylates tyrosine residues of certain proteins involved in the intracellular signaling pathways of these lymphocytes...
is recruited and phosphorylates the ITAM
ITAM
ITAM may refer to:*Instituto Tecnológico Autónomo de México, a private research university located in Mexico City, Mexico*IT asset management*Immunoreceptor tyrosine-based activation motif...
s of the CD3
CD3
CD3 or CD-3 may be:* CD3 , an antigen, cluster of differentiation protein , part of the T cell receptor complex on a mature T lymphocyte* Ford CD3 platform* MediaMax CD-3, copy protection scheme* MiniCD, a 3-inch CD...
cytoplasmic tail. ZAP70 is then recruited to the phosphorylated ITAMs and helps recruit LAT and PLC-γ
PLCG2
1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 is an enzyme that in humans is encoded by the PLCG2 gene.- Function :Enzymes of the phospholipase C family catalyze the hydrolysis of phospholipids to yield diacylglycerols and water-soluble phosphorylated derivatives of the lipid...
, which causes activation of PKC
Protein kinase C
Protein kinase C also known as PKC is a family of enzymes that are involved in controlling the function of other proteins through the phosphorylation of hydroxyl groups of serine and threonine amino acid residues on these proteins. PKC enzymes in turn are activated by signals such as increases in...
. Through a cascade of phosphorylation events, the kinase complex is activated and NF-κB is able to enter the nucleus to upregulate genes involved in T-cell development, maturation, and proliferation.
In neurons
In addition to roles in mediating cell survival, NF-κB has been demonstrated to have diverse functions in the nervous systemNervous system
The nervous system is an organ system containing a network of specialized cells called neurons that coordinate the actions of an animal and transmit signals between different parts of its body. In most animals the nervous system consists of two parts, central and peripheral. The central nervous...
including roles in plasticity
Synaptic plasticity
In neuroscience, synaptic plasticity is the ability of the connection, or synapse, between two neurons to change in strength in response to either use or disuse of transmission over synaptic pathways. Plastic change also results from the alteration of the number of receptors located on a synapse...
, learning, and memory. In addition to stimuli that activate NF-κB in other tissues, NF-κB in the nervous system can be activated by Growth Factors (BDNF, NGF
Nerve growth factor
Nerve growth factor is a small secreted protein that is important for the growth, maintenance, and survival of certain target neurons . It also functions as a signaling molecule. It is perhaps the prototypical growth factor, in that it is one of the first to be described...
) and synaptic transmission such as glutamate
Glutamic acid
Glutamic acid is one of the 20 proteinogenic amino acids, and its codons are GAA and GAG. It is a non-essential amino acid. The carboxylate anions and salts of glutamic acid are known as glutamates...
. These activators of NF-κB in the nervous system all converge upon the IKK complex and the canonical pathway.
Recently there has been a great deal of interest in the role of NF-κB in the nervous system. Current studies suggest that NF-κB is important for learning and memory in multiple organisms including crabs, fruit flies, and mice. NF-κB may regulate learning and memory in part by modulating synaptic plasticity, synapse function, as well as by regulating the growth of dendrites and dendritic spine
Dendritic spine
A dendritic spine is a small membranous protrusion from a neuron's dendrite that typically receives input from a single synapse of an axon. Dendritic spines serve as a storage site for synaptic strength and help transmit electrical signals to the neuron's cell body...
s.
Genes that have NF-κB binding sites are shown to have increased expression following learning, suggesting that the transcriptional targets of NF-κB in the nervous system are important for plasticity. Many NF-κB target genes that may be important for plasticity and learning include, glutamate receptors (AMPA-R
AMPA receptor
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor is a non-NMDA-type ionotropic transmembrane receptor for glutamate that mediates fast synaptic transmission in the central nervous system . Its name is derived from its ability to be activated by the artificial glutamate analog AMPA...
and NMDA-R
NMDA receptor
The NMDA receptor , a glutamate receptor, is the predominant molecular device for controlling synaptic plasticity and memory function....
), growth factors (BDNF, NGF) cytokines (TNF-alpha, TNFR) kinases (PKAc), and synaptic scaffolding proteins (PSD-95).
Clinical significance
Apoptosis
Apoptosis is the process of programmed cell death that may occur in multicellular organisms. Biochemical events lead to characteristic cell changes and death. These changes include blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation...
.
Defects in NF-κB results in increased susceptibility to apoptosis leading to increased cell death. This is because NF-κB regulates anti-apoptotic genes especially the TRAF1
TRAF1
TNF receptor-associated factor 1 is a protein that in humans is encoded by the TRAF1 gene.-Interactions:TRAF1 has been shown to interact with CD30, CFLAR, Baculoviral IAP repeat-containing protein 3, BIRC2, Caspase 8, HIVEP3, TNFAIP3, TRAF2, RANK and TRAF interacting protein.-Further reading:...
and TRAF2
TRAF2
TNF receptor-associated factor 2 is a protein that in humans is encoded by the TRAF2 gene.-Interactions:TRAF2 has been shown to interact with BCL10, CD30, CFLAR, IKK2, MAP3K7IP2, CD137, Caveolin 1, CD27, TNFRSF13B, TANK-binding kinase 1, TRAF1, CD40, UBE2N, MAP3K14, MAP4K2, CASP8AP2, HIVEP3,...
and, therefore, checks the activities of the caspase
Caspase
Caspases, or cysteine-aspartic proteases or cysteine-dependent aspartate-directed proteases are a family of cysteine proteases that play essential roles in apoptosis , necrosis, and inflammation....
family of enzymes, which are central to most apoptotic processes.
In tumor cells, NF-κB is active either due to mutations in genes encoding the NF-κB transcription factors themselves or in genes that control NF-κB activity (such as IκB genes); in addition, some tumor cells secrete factors that cause NF-κB to become active. Blocking NF-κB can cause tumor cells to stop proliferating, to die, or to become more sensitive to the action of anti-tumor agents. Thus, NF-κB is the subject of much active research among pharmaceutical companies as a target for anti-cancer therapy.
Because NF-κB controls many genes involved in inflammation, it is not surprising that NF-κB is found to be chronically active in many inflammatory diseases, such as inflammatory bowel disease, arthritis, sepsis, gastritis, asthma, atherosclerosis and others. It is important to note that the key regulators of NF-κB are associated with elevated mortality, especially from cardiovascular diseases. Elevated NF-κB has also been associated with schizophrenia
Schizophrenia
Schizophrenia is a mental disorder characterized by a disintegration of thought processes and of emotional responsiveness. It most commonly manifests itself as auditory hallucinations, paranoid or bizarre delusions, or disorganized speech and thinking, and it is accompanied by significant social...
.
Many natural products (including anti-oxidants) that have been promoted to have anti-cancer and anti-inflammatory activity have also been shown to inhibit NF-κB. There is a controversial US patent (US patent 6,410,516) that applies to the discovery and use of agents that can block NF-κB for therapeutic purposes. This patent is involved in several lawsuits, including Ariad v. Lilly
Ariad v. Lilly
Ariad v. Lilly is a United States court case in which Eli Lilly was found to have infringed held by Ariad Pharmaceuticals...
. Recent work by Karin, Ben-Neriah and others has highlighted the importance of the connection between NF-κB, inflammation, and cancer, and underscored the value of therapies that regulate the activity of NF-κB.
Extracts from a number of herbs and dietary plants are efficient inhibitors of NF-kappaB activation in vitro.
As a drug target
Aberrant activation of NF-κB is frequently observed in many cancers. Moreover, suppression of NF-κB limits the proliferation of cancer cells. In addition, NF-κB is a key player in the inflammatory response. Hence methods of inhibiting NF-κB signaling has potential therapeutic application in cancer and inflammatory diseases.The discovery that activation of NF-κB nuclear translocation can be separated from the elevation of oxidant stress gives an important hint to the development of strategies for NF-κB inhibition.
A new drug called denosumab
Denosumab
Denosumab is a fully human monoclonal antibody for the treatment of osteoporosis, treatment induced bone loss, bone metastases, rheumatoid arthritis, multiple myeloma and giant cell tumor of bone. It was developed by the company Amgen....
acts to raise bone mineral density and reduce fracture rates in many patient sub-groups by inhibiting RANKL
RANKL
Receptor activator of nuclear factor kappa-B ligand , also known as tumor necrosis factor ligand superfamily member 11 , TNF-related activation-induced cytokine , osteoprotegerin ligand , and osteoclast differentiation factor , is a protein that in humans is encoded by the TNFSF11 gene.RANKL is...
. RANKL acts through its receptor RANK
RANK
Receptor Activator of Nuclear Factor κ B , also known as TRANCE Receptor, is a type I membrane protein that is expressed on the surface of osteoclasts and is involved in their activation upon ligand binding...
, which in turn promotes NF-κB,
RANKL normally works by enabling the differentiation of osteoclasts from monocytes.
Disulfiram
Disulfiram
Disulfiram is a drug discovered in the 1920s and used to support the treatment of chronic alcoholism by producing an acute sensitivity to alcohol. Trade names for disulfiram in different countries are Antabuse and Antabus manufactured by Odyssey Pharmaceuticals...
, olmesartan
Olmesartan
Olmesartan medoxomil is an angiotensin II receptor antagonist used to treat high blood pressure.-Indications:...
and dithiocarbamate
Dithiocarbamate
A dithiocarbamate is a functional group in organic chemistry. It is the analog of a carbamate in which both oxygen atoms are replaced by sulfur atoms. Sodium diethyldithiocarbamate is a common ligand in inorganic chemistry....
s can inhibit the nuclear factor-κB (NF-κB) signaling cascade.
Anatabine
Anatabine
Anatabine is one of the minor alkaloids found in plants in the Solanaceae family, which includes the tobacco plant, that has been shown to affect monoamine oxidase activity...
alleged antiinflammatory effects is claimed to result from modulation of NF-κB activity.