
Glycogen storage disease type II
Encyclopedia
Glycogen storage disease type II (also called Pompe disease or acid maltase deficiency) is an autosomal recessive metabolic disorder which damages muscle and nerve cells throughout the body. It is caused by an accumulation of glycogen
in the lysosome
due to deficiency of the lysosomal acid alpha-glucosidase
enzyme. It is the only glycogen storage disease
with a defect in lysosomal metabolism, and the first glycogen storage disease to be identified, in 1932.
The build-up of glycogen causes progressive muscle weakness (myopathy
) throughout the body and affects various body tissues, particularly in the heart
, skeletal muscle
s, liver
and nervous system
.
Infantile-onset form is usually diagnosed at 4-8 months; muscles appear normal but are limp and weak preventing them from lifting their head or rolling over. As the disease progresses heart muscles thicken and progressively fail. Without treatment death usually occurs due to heart failure and respiratory weakness.
Late/later onset form occurs later than one to two years and progresses more slowly than Infantile-onset form. One of the first symptoms is a progressive decrease in muscle strength starting with the legs and moving to smaller muscles in the trunk and arms, such as the diaphragm and other muscles required for breathing. Respiratory failure is the most common cause of death. Enlargement of the heart muscles and rhythm disturbances are not significant features but do occur in some cases.
The main clinical findings include floppy baby appearance, delayed motor milestones and feeding difficulties. Moderate hepatomegaly may be present. Facial features include macroglossia, open mouth, wide open eyes, nasal flaring (due to respiratory distress), and poor facial muscle tone. Cardiopulmonary involvement is manifest by increased respiratory rate, use of accessory muscles for respiration, recurrent chest infections, decreased air entry in the left lower zone (due to cardiomegaly), arrhythmias and evidence of heart failure.
Median age at death in untreated cases is 8.7 months and is usually due to cardiorespiratory failure.
The usual initial investigations of this form of the disease include chest X ray, electrocardiogram
and echocardiography
. Typical findings are those of an enlarged heart with non specific conduction defects. Biochemical investigations include serum
creatine kinase
(typically increased 10 fold) with lesser elevations of the serum aldolase
, aspartate transaminase
, alanine transaminase
and lactic dehydrogenase. Diagnosis is made by estimating the acid alpha glucosidase activity in either skin biopsy (fibroblast
s), muscle biopsy (muscle cells) or in white blood cells. The choice of sample depends on the facilities available at the diagnostic laboratory.
Late onset features include impaired cough
, recurrent chest infections, hypotonia
, progressive muscle weakness, delayed motor milestones, difficulty swallowing or chewing and reduced vital capacity.
Prognosis depends on the age of onset on symptoms with a better prognosis being associated with later onset disease.
The findings on investigation are similar to those of the infantile form with the caveat that the creatinine kinases may be normal in some cases. The diagnosis is by estimation of the enzyme activity in a suitable sample.
(acid alpha-glucosidase
: also known as acid maltase) on long arm of chromosome 17 at 17q25.2-q25.3 (base pair 75,689,876 to 75,708,272). The number of mutations described is currently (in 2010) 289 with 67 being non-pathogenic mutations and 197 pathogenic mutations. The remainder are still being evaluated for their association with disease.
The gene spans approximately 20 kb and contains 20 exons with first exon being noncoding. The coding sequence of the putative catalytic site domain is interrupted in the middle by an intron of 101 bp. The promoter has features characteristic of a 'housekeeping' gene. The GC content is high (80%) and distinct TATA and CCAAT motifs are lacking.
Most cases appear to be due to three mutations. A transversion
(T → G) mutation is the most common among adults with this disorder. This mutation interrupts a site of RNA splicing
.
The gene encodes a protein - acid alpha-glucosidase
(EC 3.2.1.20) - which is a lysosomal hydrolase
. The protein is an enzyme
that normally degrades the alpha -1,4 and alpha -1,6 linkages in glycogen
, maltose
and isomaltose
and is required for the degradation of 1-3% of cellular glycogen
. The deficiency of this enzyme results in the accumulation of structurally normal glycogen in lysosome
s and cytoplasm
in affected individuals. Excessive glycogen storage within lysosomes may interrupt normal functioning of other organelle
s and lead to cellular injury.
A putative homologue - acid alpha-glucosidase-related gene 1 - has been identified in the nematode
Caenorhabditis elegans
.
On April 28, 2006 the US Food and Drug Administration
approved a Biologic License Application
(BLA) for Myozyme (alglucosidase alfa, rhGAA), the first treatment for patients with Pompe disease, developed by a team of Duke University
researchers. This was based on enzyme replacement therapy using biologically active recombinant human alglucosidase alfa produced in Chinese Hamster Ovary cells. Myozyme falls under the FDA Orphan Drug
designation and was approved under a priority review.
The FDA has approved Myozyme for administration by intravenous infusion of the solution. The safety and efficacy of Myozyme were assessed in two separate clinical trials in 39 infantile-onset patients with Pompe disease ranging in age from 1 month to 3.5 years at the time of the first infusion. Myozyme treatment clearly prolongs ventilator-free survival and overall survival. Early diagnosis and early treatment leads to much better outcomes. The treatment is not without side effects which include fever, flushing, skin rash, increased heart rate and even shock; these conditions, however, are usually manageable.
Myozyme costs an average of $300,000 a year, and must be taken for the patients' entire life. Some American insurers have refused to pay for it. On August 14, 2006, Health Canada approved Myozyme for the treatment of Pompe disease. On June 14, 2007 the Canadian Common Drug Review issued their recommendations regarding public funding for Myozyme therapy. Their recommendation was to provide funding to treat a very small subset of Pompe patients (Infants less one year of age with cardiomyopathy). The vast majority of developed countries are providing access to therapy for all diagnosed Pompe patients. On May 26, 2010 FDA approved Lumizyme, a similar version of Myozyme, for the treament of late-onset Pompe disease.
A new treatment option for this disease is called Lumizyme. Lumizyme and Myozyme have the same generic ingredient (Alglucosidase Alfa) and manufacturer (Genzyme Corporation). The difference between these two products is in the manufacturing process. Today, the Myozyme is made using a 160-L bioreactor, while the Lumizyme uses a 4000-L bioreactor. Because of the difference in the manufacturing process, the FDA claims that the two products are biologically different. Moreover, Lumizyme is FDA approved as replacement therapy for late-onset (noninfantile) Pompe disease without evidence of cardiac hypertrophy in patients 8 years and older. Myozyme is FDA approved for replacement therapy for infantile-onset Pompe disease.
Myozyme (alglucosidase alfa), which helps break down glucose, is a recombinant form of the human enzyme acid alpha-glucosidase
, and is also currently being used to replace the missing enzyme. In a study which included the largest cohort of patients with Pompe disease treated with enzyme replacement therapy (ERT) to date findings showed that Myozyme treatment clearly prolongs ventilator-free survival and overall survival in patients with infantile-onset Pompe disease as compared to an untreated historical control population. Furthermore, the study demonstrated that initiation of ERT prior to 6 months of age, which could be facilitated by newborn screening, shows great promise to reduce the mortality and disability associated with this devastating disorder. Taiwan and several states in the United States have started the newborn screening and results of such regimen in early diagnosis and early initiation of the therapy have dramatically improved the outcome of the disease; many of these babies have reached the normal motor developmental milestones.
Another factor affecting the treatment response is generation of antibodies against the infused enzyme, which is particularly severe in Pompe infants who have complete deficiency of the acid alpha-glucosidase. Immune tolerance therapy to eliminate these antibodies has improved the treatment outcome.
A Late Onset Treatment Study (LOTS) was published in 2010. The study was undertaken to evaluate the safety and efficacy of aglucosidase alfa in juvenile and adult patients with Pompe disease. LOTS was a randomized, double-blind, placebo-controlled study that enrolled 90 patients at eight primary sites in the United States and Europe. Participants received either aglucosidase alfa or a placebo every other week for 18 months. The average age of study participants was 44 years. The primary efficacy endpoints of the study sought to determine the effect of Myozyme on functional endurance as measured by the six-minute walk test and to determine the effect of aglucosidase alfa on pulmonary function as measured by percent predicted forced vital capacity.
The results showed that, at 78 weeks, patients treated with aglucosidase alfa increased their distance walked in six minutes by an average of approximately 25 meters as compared with the placebo group which declined by 3 meters (P=0.03). The placebo group did not show any improvement from baseline. The average baseline distance walked in six minutes in both groups was approximately 325 meters.
Percent predicted forced vital capacity in the group of patients treated with aglucosidase alfa increased by 1.2 percent at 78 weeks. In contrast, it declined by approximately 2.2 percent in the placebo group (P=0.006).
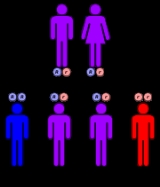
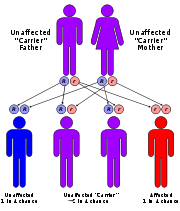 The incidence
The incidence
of the disease is approximately 1 in 140,000 for infantile GSD II and 1 in 60,000 for adult GSD II. It has been reported in almost all ethnic populations. It has an autosomal recessive inheritance pattern. This means the defective gene is located on an autosome
, and two copies of the gene—one from each parent—are required to be born with the disorder. As with all cases of autosomal recessive inheritance, children have a 1 in 4 chance of inheriting the disorder when both parents carry the defective gene, and although both parents carry one copy of the defective gene, they are usually not affected by the disorder.
, who characterized it in 1932. Pompe described accumulation of glycogen in muscle tissue in some cases of a previously unknown disorder. This accumulation was difficult to explain as the enzymes involved in the usual metabolism of glucose and glycogen were all present and functioning.
The basis for the disease remained a puzzle until Christian de Duve
's discovery of lysosomes in 1955 for which he won the Nobel Prize
in 1974. His co-worker Henri G. Hers
realised in 1965 that the deficiency of a lysosomal enzyme (alpha glucosidase) for the breakdown of glycogen could explain the symptoms of Pompe disease. This discovery led to establishing the concept of lysosomal storage diseases, of which 49 have been described (to date).
Despite recognizing the basis for the disease treatment proved difficult. Administration of the enzyme lead to its uptake by the liver and not the muscle cells where it is needed. In the early 1990s two Dutch scientists, Arnold Reuser and Ans van der Ploeg a PhD student using alpha-glucosidase containing phosphorylated mannose residues purified from bovine testes, they were able to show that an increase in the enzyme's activity in the normal mouse muscles.
Later in 1998, Dr. Yuan-Tsong Chen and colleagues at Duke University (陳垣崇,Dr. Chen is currently the director of the Institute of Biomedical Sciences, Academia Sinica
, Taipei
, Taiwan
.), using the enzyme produced in Chinese Hamster Ovary cells demonstrated for the first time that the enzyme can clear the glycogen and improved the muscle function in Pompe disease quail. The results of the work at Dukes were impressive with one treated bird recovering to the point of being able to fly again.
This was followed by production of clinical grade alpha-glucosidase in Chinese hamster ovary (CHO) cells and in the milk of transgenic rabbits. This work eventually culminated in the start of clinical trials with the first clinical trial including 4 babies receiving enzyme from rabbit milk at Erasmus MC Sophia Children’s Hospital and 3 babies receiving enzyme grown in CHO cells at Duke University in 1999.
The currently approved Myozyme is manufactured by Genzyme Corp. in Cambridge, Massachusetts, USA. Its development was a complex process. Genzyme first partnered with Pharming Group NV who had managed to produce acid alpha-glucosidase from the milk of transgentic rabbits. They also partnered with a second group based at Duke University using Chinese hamster ovary cells. In 2001, it acquired Novazyme which was also working on this enzyme. Genzyme also had its own product (Myozyme) grown in CHO cells under development. In November 2001, Genzyme chief executive Henri Termeer organised a systematic comparison of the various potential drugs in a mouse model of Pompe disease. It was found that Duke enzyme was the most efficacious, followed by Myozyme. However, due to easier manufacture of Myozyme, work on the other products was then discontinued.
Funding for research in this field was in part provided by Muscular Dystrophy Association and Acid Maltase Deficiency Association in USA and the Association of Glycogen Storage Disorders in UK and the International Pompe Association.
John Crowley
became involved in the fund-raising efforts in 1998 after two of his children were diagnosed with Pompe's. He joined the company Novazyme in 1999, which was working on enzyme replacement treatment for Pompe's. Novazyme was sold to Genzyme in 2001 for over US$100 million. The 2010 film Extraordinary Measures
is based on Crowley's search for a cure.
Glycogen
Glycogen is a molecule that serves as the secondary long-term energy storage in animal and fungal cells, with the primary energy stores being held in adipose tissue...
in the lysosome
Lysosome
thumb|350px|Schematic of typical animal cell, showing subcellular components. [[Organelle]]s: [[nucleoli]] [[cell nucleus|nucleus]] [[ribosomes]] [[vesicle |vesicle]] rough [[endoplasmic reticulum]]...
due to deficiency of the lysosomal acid alpha-glucosidase
Acid alpha-glucosidase
Lysosomal alpha-glucosidase is an enzyme that in humans is encoded by the GAA gene. Errors in this gene cause glycogen storage disease type II .-External links:* -Further reading:...
enzyme. It is the only glycogen storage disease
Glycogen storage disease
Glycogen storage disease is the result of defects in the processing of glycogen synthesis or breakdown within muscles, liver, and other cell types. GSD has two classes of cause: genetic and acquired. Genetic GSD is caused by any inborn error of metabolism involved in these processes...
with a defect in lysosomal metabolism, and the first glycogen storage disease to be identified, in 1932.
The build-up of glycogen causes progressive muscle weakness (myopathy
Myopathy
In medicine, a myopathy is a muscular disease in which the muscle fibers do not function for any one of many reasons, resulting in muscular weakness. "Myopathy" simply means muscle disease...
) throughout the body and affects various body tissues, particularly in the heart
Heart
The heart is a myogenic muscular organ found in all animals with a circulatory system , that is responsible for pumping blood throughout the blood vessels by repeated, rhythmic contractions...
, skeletal muscle
Skeletal muscle
Skeletal muscle is a form of striated muscle tissue existing under control of the somatic nervous system- i.e. it is voluntarily controlled. It is one of three major muscle types, the others being cardiac and smooth muscle...
s, liver
Liver
The liver is a vital organ present in vertebrates and some other animals. It has a wide range of functions, including detoxification, protein synthesis, and production of biochemicals necessary for digestion...
and nervous system
Nervous system
The nervous system is an organ system containing a network of specialized cells called neurons that coordinate the actions of an animal and transmit signals between different parts of its body. In most animals the nervous system consists of two parts, central and peripheral. The central nervous...
.
Classification
There are exceptions but levels of alpha-glucosidase determines the type of GSD II an individual may have.More alpha glucosidase present in the individuals muscles means symptoms occur later in life and progress more slowly. GSD II is broadly divided into two onset forms based on the age symptoms occur.Infantile-onset form is usually diagnosed at 4-8 months; muscles appear normal but are limp and weak preventing them from lifting their head or rolling over. As the disease progresses heart muscles thicken and progressively fail. Without treatment death usually occurs due to heart failure and respiratory weakness.
Late/later onset form occurs later than one to two years and progresses more slowly than Infantile-onset form. One of the first symptoms is a progressive decrease in muscle strength starting with the legs and moving to smaller muscles in the trunk and arms, such as the diaphragm and other muscles required for breathing. Respiratory failure is the most common cause of death. Enlargement of the heart muscles and rhythm disturbances are not significant features but do occur in some cases.
Infantile
The infantile form usually comes to medical attention within the first few months of life. The usual presenting features are cardiomegaly (92%), hypotonia (88%), cardiomyopathy (88%), respiratory distress (78%), muscle weakness (63%), feeding difficulties (57%) and failure to thrive (53%).The main clinical findings include floppy baby appearance, delayed motor milestones and feeding difficulties. Moderate hepatomegaly may be present. Facial features include macroglossia, open mouth, wide open eyes, nasal flaring (due to respiratory distress), and poor facial muscle tone. Cardiopulmonary involvement is manifest by increased respiratory rate, use of accessory muscles for respiration, recurrent chest infections, decreased air entry in the left lower zone (due to cardiomegaly), arrhythmias and evidence of heart failure.
Median age at death in untreated cases is 8.7 months and is usually due to cardiorespiratory failure.
The usual initial investigations of this form of the disease include chest X ray, electrocardiogram
Electrocardiogram
Electrocardiography is a transthoracic interpretation of the electrical activity of the heart over a period of time, as detected by electrodes attached to the outer surface of the skin and recorded by a device external to the body...
and echocardiography
Echocardiography
An echocardiogram, often referred to in the medical community as a cardiac ECHO or simply an ECHO, is a sonogram of the heart . Also known as a cardiac ultrasound, it uses standard ultrasound techniques to image two-dimensional slices of the heart...
. Typical findings are those of an enlarged heart with non specific conduction defects. Biochemical investigations include serum
Serum
Serum may refer to:*Blood serum, a component of blood which is collected after coagulation.**Antiserum, blood serum with specific antibodies for passive immunity*Serous fluid, any clear bodily fluid*any drug derived from an animal's blood or serous fluid...
creatine kinase
Creatine kinase
Creatine kinase , also known as creatine phosphokinase or phospho-creatine kinase , is an enzyme expressed by various tissues and cell types. CK catalyses the conversion of creatine and consumes adenosine triphosphate to create phosphocreatine and adenosine diphosphate...
(typically increased 10 fold) with lesser elevations of the serum aldolase
Aldolase
Aldolase A is an enzyme that catalyses a reverse aldol reaction: The substrate, fructose 1,6-bisphosphate is broken down into glyceraldehyde 3-phosphate and dihydroxyacetone phosphate . This reaction is a part of glycolysis. Three aldolase isozymes , encoded by three different genes, are...
, aspartate transaminase
Aspartate transaminase
Aspartate transaminase , also called aspartate aminotransferase or serum glutamic oxaloacetic transaminase , is a pyridoxal phosphate -dependent transaminase enzyme . AST catalyzes the reversible transfer of an α-amino group between aspartate and glutamate and, as such, is an important enzyme in...
, alanine transaminase
Alanine transaminase
Alanine transaminase or ALT is a transaminase enzyme . It is also called serum glutamic pyruvic transaminase or alanine aminotransferase ....
and lactic dehydrogenase. Diagnosis is made by estimating the acid alpha glucosidase activity in either skin biopsy (fibroblast
Fibroblast
A fibroblast is a type of cell that synthesizes the extracellular matrix and collagen, the structural framework for animal tissues, and plays a critical role in wound healing...
s), muscle biopsy (muscle cells) or in white blood cells. The choice of sample depends on the facilities available at the diagnostic laboratory.
Late onset form
This form differs from the infantile principally in the relative lack of cardiac involvement. The onset is more insidious and has a slower progression. Cardiac involvement may occur but is milder than in the infantile form. Skeletal involvement is more prominent with a predilection for the lower limbs.Late onset features include impaired cough
Cough
A cough is a sudden and often repetitively occurring reflex which helps to clear the large breathing passages from secretions, irritants, foreign particles and microbes...
, recurrent chest infections, hypotonia
Hypotonia
Hypotonia is a state of low muscle tone , often involving reduced muscle strength. Hypotonia is not a specific medical disorder, but a potential manifestation of many different diseases and disorders that affect motor nerve control by the brain or muscle strength...
, progressive muscle weakness, delayed motor milestones, difficulty swallowing or chewing and reduced vital capacity.
Prognosis depends on the age of onset on symptoms with a better prognosis being associated with later onset disease.
The findings on investigation are similar to those of the infantile form with the caveat that the creatinine kinases may be normal in some cases. The diagnosis is by estimation of the enzyme activity in a suitable sample.
Cause
The disease is caused by a mutation in a geneGene
A gene is a molecular unit of heredity of a living organism. It is a name given to some stretches of DNA and RNA that code for a type of protein or for an RNA chain that has a function in the organism. Living beings depend on genes, as they specify all proteins and functional RNA chains...
(acid alpha-glucosidase
Acid alpha-glucosidase
Lysosomal alpha-glucosidase is an enzyme that in humans is encoded by the GAA gene. Errors in this gene cause glycogen storage disease type II .-External links:* -Further reading:...
: also known as acid maltase) on long arm of chromosome 17 at 17q25.2-q25.3 (base pair 75,689,876 to 75,708,272). The number of mutations described is currently (in 2010) 289 with 67 being non-pathogenic mutations and 197 pathogenic mutations. The remainder are still being evaluated for their association with disease.
The gene spans approximately 20 kb and contains 20 exons with first exon being noncoding. The coding sequence of the putative catalytic site domain is interrupted in the middle by an intron of 101 bp. The promoter has features characteristic of a 'housekeeping' gene. The GC content is high (80%) and distinct TATA and CCAAT motifs are lacking.
Most cases appear to be due to three mutations. A transversion
Transversion
In molecular biology, transversion refers to the substitution of a purine for a pyrimidine or vice versa. It can only be reverted by a spontaneous reversion. Because this type of mutation changes the chemical structure dramatically, the consequences of this change tend to be more drastic than those...
(T → G) mutation is the most common among adults with this disorder. This mutation interrupts a site of RNA splicing
RNA splicing
In molecular biology and genetics, splicing is a modification of an RNA after transcription, in which introns are removed and exons are joined. This is needed for the typical eukaryotic messenger RNA before it can be used to produce a correct protein through translation...
.
The gene encodes a protein - acid alpha-glucosidase
Acid alpha-glucosidase
Lysosomal alpha-glucosidase is an enzyme that in humans is encoded by the GAA gene. Errors in this gene cause glycogen storage disease type II .-External links:* -Further reading:...
(EC 3.2.1.20) - which is a lysosomal hydrolase
Hydrolase
In biochemistry, a hydrolase is an enzyme that catalyzes the hydrolysis of a chemical bond. For example, an enzyme that catalyzed the following reaction is a hydrolase:-Nomenclature:...
. The protein is an enzyme
Enzyme
Enzymes are proteins that catalyze chemical reactions. In enzymatic reactions, the molecules at the beginning of the process, called substrates, are converted into different molecules, called products. Almost all chemical reactions in a biological cell need enzymes in order to occur at rates...
that normally degrades the alpha -1,4 and alpha -1,6 linkages in glycogen
Glycogen
Glycogen is a molecule that serves as the secondary long-term energy storage in animal and fungal cells, with the primary energy stores being held in adipose tissue...
, maltose
Maltose
Maltose , or malt sugar, is a disaccharide formed from two units of glucose joined with an αbond, formed from a condensation reaction. The isomer "isomaltose" has two glucose molecules linked through an α bond. Maltose is the second member of an important biochemical series of glucose chains....
and isomaltose
Isomaltose
Isomaltose is a disaccharide similar to maltose, but with a α--linkage instead of the α--linkage. It is a reducing sugar. Both of the sugars are glucose and pyranoses...
and is required for the degradation of 1-3% of cellular glycogen
Glycogen
Glycogen is a molecule that serves as the secondary long-term energy storage in animal and fungal cells, with the primary energy stores being held in adipose tissue...
. The deficiency of this enzyme results in the accumulation of structurally normal glycogen in lysosome
Lysosome
thumb|350px|Schematic of typical animal cell, showing subcellular components. [[Organelle]]s: [[nucleoli]] [[cell nucleus|nucleus]] [[ribosomes]] [[vesicle |vesicle]] rough [[endoplasmic reticulum]]...
s and cytoplasm
Cytoplasm
The cytoplasm is a small gel-like substance residing between the cell membrane holding all the cell's internal sub-structures , except for the nucleus. All the contents of the cells of prokaryote organisms are contained within the cytoplasm...
in affected individuals. Excessive glycogen storage within lysosomes may interrupt normal functioning of other organelle
Organelle
In cell biology, an organelle is a specialized subunit within a cell that has a specific function, and is usually separately enclosed within its own lipid bilayer....
s and lead to cellular injury.
A putative homologue - acid alpha-glucosidase-related gene 1 - has been identified in the nematode
Nematode
The nematodes or roundworms are the most diverse phylum of pseudocoelomates, and one of the most diverse of all animals. Nematode species are very difficult to distinguish; over 28,000 have been described, of which over 16,000 are parasitic. It has been estimated that the total number of nematode...
Caenorhabditis elegans
Caenorhabditis elegans
Caenorhabditis elegans is a free-living, transparent nematode , about 1 mm in length, which lives in temperate soil environments. Research into the molecular and developmental biology of C. elegans was begun in 1974 by Sydney Brenner and it has since been used extensively as a model...
.
Treatment
Cardiac and respiratory complications are treated symptomatically. Physical and occupational therapy may be beneficial for some patients. Alterations in diet may provide temporary improvement but will not alter the course of the disease. Genetic counseling can provide families with information regarding risk in future pregnancies.On April 28, 2006 the US Food and Drug Administration
Food and Drug Administration
The Food and Drug Administration is an agency of the United States Department of Health and Human Services, one of the United States federal executive departments...
approved a Biologic License Application
Biologic License Application
As defined by the US FDA, a Biologic License Application is: Biological products are approved for marketing under the provisions of the Public Health Service Act. The Act requires a firm who manufactures a biologic for sale in interstate commerce to hold a license for the product...
(BLA) for Myozyme (alglucosidase alfa, rhGAA), the first treatment for patients with Pompe disease, developed by a team of Duke University
Duke University
Duke University is a private research university located in Durham, North Carolina, United States. Founded by Methodists and Quakers in the present day town of Trinity in 1838, the school moved to Durham in 1892. In 1924, tobacco industrialist James B...
researchers. This was based on enzyme replacement therapy using biologically active recombinant human alglucosidase alfa produced in Chinese Hamster Ovary cells. Myozyme falls under the FDA Orphan Drug
Orphan drug
An orphan drug is a pharmaceutical agent that has been developed specifically to treat a rare medical condition, the condition itself being referred to as an orphan disease...
designation and was approved under a priority review.
The FDA has approved Myozyme for administration by intravenous infusion of the solution. The safety and efficacy of Myozyme were assessed in two separate clinical trials in 39 infantile-onset patients with Pompe disease ranging in age from 1 month to 3.5 years at the time of the first infusion. Myozyme treatment clearly prolongs ventilator-free survival and overall survival. Early diagnosis and early treatment leads to much better outcomes. The treatment is not without side effects which include fever, flushing, skin rash, increased heart rate and even shock; these conditions, however, are usually manageable.
Myozyme costs an average of $300,000 a year, and must be taken for the patients' entire life. Some American insurers have refused to pay for it. On August 14, 2006, Health Canada approved Myozyme for the treatment of Pompe disease. On June 14, 2007 the Canadian Common Drug Review issued their recommendations regarding public funding for Myozyme therapy. Their recommendation was to provide funding to treat a very small subset of Pompe patients (Infants less one year of age with cardiomyopathy). The vast majority of developed countries are providing access to therapy for all diagnosed Pompe patients. On May 26, 2010 FDA approved Lumizyme, a similar version of Myozyme, for the treament of late-onset Pompe disease.
A new treatment option for this disease is called Lumizyme. Lumizyme and Myozyme have the same generic ingredient (Alglucosidase Alfa) and manufacturer (Genzyme Corporation). The difference between these two products is in the manufacturing process. Today, the Myozyme is made using a 160-L bioreactor, while the Lumizyme uses a 4000-L bioreactor. Because of the difference in the manufacturing process, the FDA claims that the two products are biologically different. Moreover, Lumizyme is FDA approved as replacement therapy for late-onset (noninfantile) Pompe disease without evidence of cardiac hypertrophy in patients 8 years and older. Myozyme is FDA approved for replacement therapy for infantile-onset Pompe disease.
Prognosis
The prognosis for individuals with Pompe disease varies according to the onset and severity of symptoms. Without treatment the disease is particularly lethal in infants and young children.Myozyme (alglucosidase alfa), which helps break down glucose, is a recombinant form of the human enzyme acid alpha-glucosidase
Acid alpha-glucosidase
Lysosomal alpha-glucosidase is an enzyme that in humans is encoded by the GAA gene. Errors in this gene cause glycogen storage disease type II .-External links:* -Further reading:...
, and is also currently being used to replace the missing enzyme. In a study which included the largest cohort of patients with Pompe disease treated with enzyme replacement therapy (ERT) to date findings showed that Myozyme treatment clearly prolongs ventilator-free survival and overall survival in patients with infantile-onset Pompe disease as compared to an untreated historical control population. Furthermore, the study demonstrated that initiation of ERT prior to 6 months of age, which could be facilitated by newborn screening, shows great promise to reduce the mortality and disability associated with this devastating disorder. Taiwan and several states in the United States have started the newborn screening and results of such regimen in early diagnosis and early initiation of the therapy have dramatically improved the outcome of the disease; many of these babies have reached the normal motor developmental milestones.
Another factor affecting the treatment response is generation of antibodies against the infused enzyme, which is particularly severe in Pompe infants who have complete deficiency of the acid alpha-glucosidase. Immune tolerance therapy to eliminate these antibodies has improved the treatment outcome.
A Late Onset Treatment Study (LOTS) was published in 2010. The study was undertaken to evaluate the safety and efficacy of aglucosidase alfa in juvenile and adult patients with Pompe disease. LOTS was a randomized, double-blind, placebo-controlled study that enrolled 90 patients at eight primary sites in the United States and Europe. Participants received either aglucosidase alfa or a placebo every other week for 18 months. The average age of study participants was 44 years. The primary efficacy endpoints of the study sought to determine the effect of Myozyme on functional endurance as measured by the six-minute walk test and to determine the effect of aglucosidase alfa on pulmonary function as measured by percent predicted forced vital capacity.
The results showed that, at 78 weeks, patients treated with aglucosidase alfa increased their distance walked in six minutes by an average of approximately 25 meters as compared with the placebo group which declined by 3 meters (P=0.03). The placebo group did not show any improvement from baseline. The average baseline distance walked in six minutes in both groups was approximately 325 meters.
Percent predicted forced vital capacity in the group of patients treated with aglucosidase alfa increased by 1.2 percent at 78 weeks. In contrast, it declined by approximately 2.2 percent in the placebo group (P=0.006).
Epidemiology
Incidence
Incidence may refer to:* Incidence , a measure of the risk of developing some new condition within a specified period of time* Incidence , the binary relations describing how subsets meet...
of the disease is approximately 1 in 140,000 for infantile GSD II and 1 in 60,000 for adult GSD II. It has been reported in almost all ethnic populations. It has an autosomal recessive inheritance pattern. This means the defective gene is located on an autosome
Autosome
An autosome is a chromosome that is not a sex chromosome, or allosome; that is to say, there is an equal number of copies of the chromosome in males and females. For example, in humans, there are 22 pairs of autosomes. In addition to autosomes, there are sex chromosomes, to be specific: X and Y...
, and two copies of the gene—one from each parent—are required to be born with the disorder. As with all cases of autosomal recessive inheritance, children have a 1 in 4 chance of inheriting the disorder when both parents carry the defective gene, and although both parents carry one copy of the defective gene, they are usually not affected by the disorder.
History
The disease is named after Johann PompeJohann Pompe
Joannes Cassianus Pompe was a Dutch pathologist.He characterized the condition now known as Glycogen storage disease type II in 1932. It is sometimes referred to by his name.He was executed by the German army in April 1945 for espionage....
, who characterized it in 1932. Pompe described accumulation of glycogen in muscle tissue in some cases of a previously unknown disorder. This accumulation was difficult to explain as the enzymes involved in the usual metabolism of glucose and glycogen were all present and functioning.
The basis for the disease remained a puzzle until Christian de Duve
Christian de Duve
Christian René, viscount de Duve is a Nobel Prize-winning cytologist and biochemist. De Duve was born in Thames Ditton, Surrey, Great Britain, as a son of Belgian refugees. They returned to Belgium in 1920...
's discovery of lysosomes in 1955 for which he won the Nobel Prize
Nobel Prize
The Nobel Prizes are annual international awards bestowed by Scandinavian committees in recognition of cultural and scientific advances. The will of the Swedish chemist Alfred Nobel, the inventor of dynamite, established the prizes in 1895...
in 1974. His co-worker Henri G. Hers
Henri G. Hers
Henri-Géry Hers is a Belgian physiologist and biochemist, and was a professor at the Universite Catholique de Louvain. Hers' suffers from an inborn glycogen metabolism disorder caused by deficiency of hepatic phosphorylase associated with an enlarged liver and mild hypoglycaemia...
realised in 1965 that the deficiency of a lysosomal enzyme (alpha glucosidase) for the breakdown of glycogen could explain the symptoms of Pompe disease. This discovery led to establishing the concept of lysosomal storage diseases, of which 49 have been described (to date).
Despite recognizing the basis for the disease treatment proved difficult. Administration of the enzyme lead to its uptake by the liver and not the muscle cells where it is needed. In the early 1990s two Dutch scientists, Arnold Reuser and Ans van der Ploeg a PhD student using alpha-glucosidase containing phosphorylated mannose residues purified from bovine testes, they were able to show that an increase in the enzyme's activity in the normal mouse muscles.
Later in 1998, Dr. Yuan-Tsong Chen and colleagues at Duke University (陳垣崇,Dr. Chen is currently the director of the Institute of Biomedical Sciences, Academia Sinica
Academia Sinica
The Academia Sinica , headquartered in the Nangang District of Taipei, is the national academy of Taiwan. It supports research activities in a wide variety of disciplines, ranging from mathematical and physical sciences, to life sciences, and to humanities and social sciences.Academia Sinica has...
, Taipei
Taipei
Taipei City is the capital of the Republic of China and the central city of the largest metropolitan area of Taiwan. Situated at the northern tip of the island, Taipei is located on the Tamsui River, and is about 25 km southwest of Keelung, its port on the Pacific Ocean...
, Taiwan
Taiwan
Taiwan , also known, especially in the past, as Formosa , is the largest island of the same-named island group of East Asia in the western Pacific Ocean and located off the southeastern coast of mainland China. The island forms over 99% of the current territory of the Republic of China following...
.), using the enzyme produced in Chinese Hamster Ovary cells demonstrated for the first time that the enzyme can clear the glycogen and improved the muscle function in Pompe disease quail. The results of the work at Dukes were impressive with one treated bird recovering to the point of being able to fly again.
This was followed by production of clinical grade alpha-glucosidase in Chinese hamster ovary (CHO) cells and in the milk of transgenic rabbits. This work eventually culminated in the start of clinical trials with the first clinical trial including 4 babies receiving enzyme from rabbit milk at Erasmus MC Sophia Children’s Hospital and 3 babies receiving enzyme grown in CHO cells at Duke University in 1999.
The currently approved Myozyme is manufactured by Genzyme Corp. in Cambridge, Massachusetts, USA. Its development was a complex process. Genzyme first partnered with Pharming Group NV who had managed to produce acid alpha-glucosidase from the milk of transgentic rabbits. They also partnered with a second group based at Duke University using Chinese hamster ovary cells. In 2001, it acquired Novazyme which was also working on this enzyme. Genzyme also had its own product (Myozyme) grown in CHO cells under development. In November 2001, Genzyme chief executive Henri Termeer organised a systematic comparison of the various potential drugs in a mouse model of Pompe disease. It was found that Duke enzyme was the most efficacious, followed by Myozyme. However, due to easier manufacture of Myozyme, work on the other products was then discontinued.
Funding for research in this field was in part provided by Muscular Dystrophy Association and Acid Maltase Deficiency Association in USA and the Association of Glycogen Storage Disorders in UK and the International Pompe Association.
John Crowley
John Crowley (biotech executive)
John Francis Crowley is an American biotechnology executive and entrepreneur. He is best known as the founder of several biotech companies devoted to curing genetic diseases.-Life and career:...
became involved in the fund-raising efforts in 1998 after two of his children were diagnosed with Pompe's. He joined the company Novazyme in 1999, which was working on enzyme replacement treatment for Pompe's. Novazyme was sold to Genzyme in 2001 for over US$100 million. The 2010 film Extraordinary Measures
Extraordinary Measures
Extraordinary Measures is a 2010 medical drama film starring Brendan Fraser, Harrison Ford, and Keri Russell. It is distributed by CBS Films and was released on January 22, 2010. It is about parents who form a biotechnology company to develop a drug to save the lives of their children, who have a...
is based on Crowley's search for a cure.
External links
- The website of the Pompe's Group of the Association for Glycogen Storage Disease (UK)
- Acid Maltase Deficiency Association, Inc.—A US patient support group founded in 1995.
- United Pompe Foundation
- International Pompe Association—A federation of Pompe disease patient's groups worldwide.
- Genzyme's Pompe Information site
- FDA Approval News for Myozyme
- Canadian Association of Pompe
- Hide & Seek Foundation for Lysosomal Disease Research
- GeneReview/NIH/UW entry on Glycogen Storage Disease Type II (Pompe Disease)
- Australian Pompe's Association