
Langerhans cell histiocytosis
Encyclopedia
Langerhans cell Histiocytosis (LCH) is a rare disease
involving clonal proliferation of Langerhans cell
s, abnormal cell
s deriving from bone marrow
and capable of migrating from skin to lymph node
s. Clinically, its manifestations range from isolated bone lesions to multisystem disease
.
LCH is part of a group of clinical syndromes called histiocytoses
, which are characterized by an abnormal proliferation of histiocyte
s (an archaic term for activated dendritic cell
s and macrophage
s). These diseases are related to other forms of abnormal proliferation of white blood cell
s, such as leukemia
s and lymphoma
s.
The disease has gone by several names, including Hand-Schüller-Christian disease, Abt-Letterer-Siwe disease, and histiocytosis X, until it was renamed in 1985 by the Histiocyte Society.
The disease spectrum results from clonal accumulation and proliferation
of cells resembling the epidermal dendritic cell
s called Langerhans cell
s, hence sometimes called dendritic cell histiocytosis. These cells in combination with lymphocyte
s, eosinophils, and normal histiocyte
s form typical LCH lesions that can be found in almost any organ
.
There are three types of histiocytoses: malignant (true histiocytic lymphomas), "reactive" (benign histiocytoses), Langerhans cell histiocytosis.
"Reactive" in this context indicates that the abnormality may be due to a physiological reaction to infection. For example leukocytosis
(proliferation of white blood cells) is a normal reaction to infection, and "histiocytes" are developmentally related to white blood cells.
LCH is traditionally divided into three groups: unifocal, multifocal unisystem, and multifocal multisystem.
Unifocal
Multifocal unisystem
Multifocal multisystem
Pulmonary Langerhans cell histiocytosis (PLCH) is a unique form of LCH in that it occurs almost exclusively in cigarette smokers. It is now considered a form of smoking-related interstitial lung disease. Some patients recover completely after they stop smoking, but others develop long-term complications such as pulmonary fibrosis
and pulmonary hypertension
.
LCH is usually a sporadic and non-hereditary condition but familial
clustering has been noted in limited number of cases. Hashimoto-Pritzker disease is a congenital self-healing variant of Hand-Schüller-Christian disease.
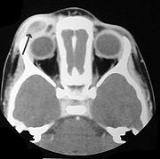
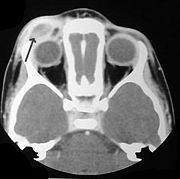 LCH provokes a non-specific inflammatory response
LCH provokes a non-specific inflammatory response
, which includes fever, lethargy, and weight loss. Organ involvement can also cause more specific symptoms.
biopsy
. Hematoxylin-eosin stain of biopsy slide will show features of Langerhans cell e.g. distinct cell margin, pink granular cytoplasm
. Presence of Birbeck granules
on electron microscopy and immuno-cytochemical
features e. g. CD1
positivity are more specific. Initially routine blood tests e.g. full blood count, liver function test, U&Es, bone profile are done to determine disease extent and rule out other causes. Radiology will show osteolytic bone lesions and damage to the lung. The latter may be evident in chest X-ray
s with micronodular and interstitial
infiltrate in the mid and lower zone of lung, with sparing of the Costophrenic angle
or honeycomb
appearance in older lesions. MRI and CT
may show infiltration in sella turcica
. Assessment of endocrine function and bonemarrow biopsy are also performed when indicated.
for diabetes insipidus which can be applied as nasal drop. Chemotherapeutic agents such as alkylating agents
, antimetabolites, vinca alkaloids
either singly or in combination can lead to complete remission in diffuse disease.
Disease
A disease is an abnormal condition affecting the body of an organism. It is often construed to be a medical condition associated with specific symptoms and signs. It may be caused by external factors, such as infectious disease, or it may be caused by internal dysfunctions, such as autoimmune...
involving clonal proliferation of Langerhans cell
Langerhans cell
Langerhans cells are dendritic cells of the skin and mucosa, and contain large granules called Birbeck granules. They are present in all layers of the epidermis, but are most prominant in the stratum spinosum. They also occur in the papillary dermis, particularly around blood vessels, as well as...
s, abnormal cell
Cell (biology)
The cell is the basic structural and functional unit of all known living organisms. It is the smallest unit of life that is classified as a living thing, and is often called the building block of life. The Alberts text discusses how the "cellular building blocks" move to shape developing embryos....
s deriving from bone marrow
Bone marrow
Bone marrow is the flexible tissue found in the interior of bones. In humans, bone marrow in large bones produces new blood cells. On average, bone marrow constitutes 4% of the total body mass of humans; in adults weighing 65 kg , bone marrow accounts for approximately 2.6 kg...
and capable of migrating from skin to lymph node
Lymph node
A lymph node is a small ball or an oval-shaped organ of the immune system, distributed widely throughout the body including the armpit and stomach/gut and linked by lymphatic vessels. Lymph nodes are garrisons of B, T, and other immune cells. Lymph nodes are found all through the body, and act as...
s. Clinically, its manifestations range from isolated bone lesions to multisystem disease
Systemic disease
Life-threatening disease redirects here.A systemic disease is one that affects a number of organs and tissues, or affects the body as a whole. Although most medical conditions will eventually involve multiple organs in advanced stage Life-threatening disease redirects here.A systemic disease is one...
.
LCH is part of a group of clinical syndromes called histiocytoses
Histiocytosis
In medicine, histiocytosis refers to an excessive number of histiocytes, , and is typically used to refer to a group of rare diseases which share this as a characteristic...
, which are characterized by an abnormal proliferation of histiocyte
Histiocyte
A histiocyte is a cell that is part of the mononuclear phagocyte system . The mononuclear phagocytic system is part of the organism's immune system...
s (an archaic term for activated dendritic cell
Dendritic cell
Dendritic cells are immune cells forming part of the mammalian immune system. Their main function is to process antigen material and present it on the surface to other cells of the immune system. That is, dendritic cells function as antigen-presenting cells...
s and macrophage
Macrophage
Macrophages are cells produced by the differentiation of monocytes in tissues. Human macrophages are about in diameter. Monocytes and macrophages are phagocytes. Macrophages function in both non-specific defense as well as help initiate specific defense mechanisms of vertebrate animals...
s). These diseases are related to other forms of abnormal proliferation of white blood cell
White blood cell
White blood cells, or leukocytes , are cells of the immune system involved in defending the body against both infectious disease and foreign materials. Five different and diverse types of leukocytes exist, but they are all produced and derived from a multipotent cell in the bone marrow known as a...
s, such as leukemia
Leukemia
Leukemia or leukaemia is a type of cancer of the blood or bone marrow characterized by an abnormal increase of immature white blood cells called "blasts". Leukemia is a broad term covering a spectrum of diseases...
s and lymphoma
Lymphoma
Lymphoma is a cancer in the lymphatic cells of the immune system. Typically, lymphomas present as a solid tumor of lymphoid cells. Treatment might involve chemotherapy and in some cases radiotherapy and/or bone marrow transplantation, and can be curable depending on the histology, type, and stage...
s.
The disease has gone by several names, including Hand-Schüller-Christian disease, Abt-Letterer-Siwe disease, and histiocytosis X, until it was renamed in 1985 by the Histiocyte Society.
Classification
| Alternative names |
|---|
| Histiocytosis X Histiocytosis X syndrome |
| Subordinate terms |
| Hand-Schüller-Christian disease Letterer-Siwe disease Histiocytosis X, unspecified Eosinophilic granulomatosis Langerhans cell granulomatosis Langerhans cell histiocytosis, Hashimoto-Pritzker type Langerhans cell histiocytosis of lung Langerhans cell histiocytosis, disseminated (clinical) Langerhans cell histiocytosis, unifocal (clinical) |
The disease spectrum results from clonal accumulation and proliferation
Cell growth
The term cell growth is used in the contexts of cell development and cell division . When used in the context of cell division, it refers to growth of cell populations, where one cell grows and divides to produce two "daughter cells"...
of cells resembling the epidermal dendritic cell
Dendritic cell
Dendritic cells are immune cells forming part of the mammalian immune system. Their main function is to process antigen material and present it on the surface to other cells of the immune system. That is, dendritic cells function as antigen-presenting cells...
s called Langerhans cell
Langerhans cell
Langerhans cells are dendritic cells of the skin and mucosa, and contain large granules called Birbeck granules. They are present in all layers of the epidermis, but are most prominant in the stratum spinosum. They also occur in the papillary dermis, particularly around blood vessels, as well as...
s, hence sometimes called dendritic cell histiocytosis. These cells in combination with lymphocyte
Lymphocyte
A lymphocyte is a type of white blood cell in the vertebrate immune system.Under the microscope, lymphocytes can be divided into large lymphocytes and small lymphocytes. Large granular lymphocytes include natural killer cells...
s, eosinophils, and normal histiocyte
Histiocyte
A histiocyte is a cell that is part of the mononuclear phagocyte system . The mononuclear phagocytic system is part of the organism's immune system...
s form typical LCH lesions that can be found in almost any organ
Organ (anatomy)
In biology, an organ is a collection of tissues joined in structural unit to serve a common function. Usually there is a main tissue and sporadic tissues . The main tissue is the one that is unique for the specific organ. For example, main tissue in the heart is the myocardium, while sporadic are...
.
There are three types of histiocytoses: malignant (true histiocytic lymphomas), "reactive" (benign histiocytoses), Langerhans cell histiocytosis.
"Reactive" in this context indicates that the abnormality may be due to a physiological reaction to infection. For example leukocytosis
Leukocytosis
Leukocytosis is a raised white blood cell count above the normal range in the blood. It is frequently a sign of an inflammatory response, most commonly the result of infection, and is observed in certain parasitic infections...
(proliferation of white blood cells) is a normal reaction to infection, and "histiocytes" are developmentally related to white blood cells.
LCH is traditionally divided into three groups: unifocal, multifocal unisystem, and multifocal multisystem.
Unifocal
- Unifocal LCH, also called eosinophilic granuloma (an older term which is now known to be a misnomerMisnomerA misnomer is a term which suggests an interpretation that is known to be untrue. Such incorrect terms sometimes derive their names because of the form, action, or origin of the subject becoming named popularly or widely referenced—long before their true natures were known.- Sources of misnomers...
), is a slowly-progressing disease characterized by an expanding proliferation of Langerhans cells in various bones. It is a monostotic or polystotic disease with no extraskeletal involvement. This differentiates eosinofilic granuloma from other forms of langerhans cell histiocytosis (Letterer-Siwe or Hand-Schüller-Christian variant.
Multifocal unisystem
- Seen mostly in children, multifocal unisystem LCH is characterized by fever, bone lesions and diffuse eruptions, usually on the scalp and in the ear canals. 50% of cases involve the pituitary stalkPituitary glandIn vertebrate anatomy the pituitary gland, or hypophysis, is an endocrine gland about the size of a pea and weighing 0.5 g , in humans. It is a protrusion off the bottom of the hypothalamus at the base of the brain, and rests in a small, bony cavity covered by a dural fold...
, leading to diabetes insipidusDiabetes insipidusDiabetes insipidus is a condition characterized by excessive thirst and excretion of large amounts of severely diluted urine, with reduction of fluid intake having no effect on the concentration of the urine. There are several different types of DI, each with a different cause...
. The triad of diabetes insipidus, exopthalmos, and lytic bone lesions is known as the Hand-Schüller-Christian triad.
Multifocal multisystem
- Multifocal multisystem LCH, also called Letterer-Siwe diseaseLetterer-Siwe diseaseLetterer–Siwe disease is a genetic disorder considered to be a type of histiocytosis . It is sometimes classified as a form of Langerhans cell histiocytosis, or as a form of histiocytosis X. It is most commonly seen in children less than two years old...
, is a rapidly-progressing disease in which Langerhans cells proliferate in many tissues. It is mostly seen in children under age 2, and the prognosis is poor: even with aggressive chemotherapy, the 5-year survival is only 50%.
Pulmonary Langerhans cell histiocytosis (PLCH) is a unique form of LCH in that it occurs almost exclusively in cigarette smokers. It is now considered a form of smoking-related interstitial lung disease. Some patients recover completely after they stop smoking, but others develop long-term complications such as pulmonary fibrosis
Pulmonary fibrosis
Pulmonary fibrosis is the formation or development of excess fibrous connective tissue in the lungs. It is also described as "scarring of the lung".-Symptoms:Symptoms of pulmonary fibrosis are mainly:...
and pulmonary hypertension
Pulmonary hypertension
In medicine, pulmonary hypertension is an increase in blood pressure in the pulmonary artery, pulmonary vein, or pulmonary capillaries, together known as the lung vasculature, leading to shortness of breath, dizziness, fainting, and other symptoms, all of which are exacerbated by exertion...
.
Prevalence
LCH usually affects children between 1 and 15 years old, with a peak incidence between 5 and 10 years of age. Among children under the age of 10, yearly incidence is thought to be 1 in 200,000; and in adults even rarer, in about 1 in 560,000. It has been reported in elderly but is vanishingly rare. It is most prevalent in Caucasians, and affects males twice as often as females.LCH is usually a sporadic and non-hereditary condition but familial
Family
In human context, a family is a group of people affiliated by consanguinity, affinity, or co-residence. In most societies it is the principal institution for the socialization of children...
clustering has been noted in limited number of cases. Hashimoto-Pritzker disease is a congenital self-healing variant of Hand-Schüller-Christian disease.
Signs and symptoms
Inflammation
Inflammation is part of the complex biological response of vascular tissues to harmful stimuli, such as pathogens, damaged cells, or irritants. Inflammation is a protective attempt by the organism to remove the injurious stimuli and to initiate the healing process...
, which includes fever, lethargy, and weight loss. Organ involvement can also cause more specific symptoms.
- Bone: The most-frequently seen symptom in both unifocal and multifocal disease is painful bone swelling. The skull is most frequently affected, followed by the long bones of the upper extremities and flat bones. Infiltration in hands and feet is unusual. Osteolytic lesions can lead to pathological fractures.
- Skin: Commonly seen are a rash which varies from scaly erythematous lesions to red papules pronounced in intertriginousIntertrigoAn intertrigo is an inflammation of the body folds .An intertrigo sometimes refers to a bacterial, fungal, or viral infection that has developed at the site of broken skin due to such inflammation...
areas. Up to 80% of LCH patients have extensive eruptions on the scalp. - Bone marrow: Pancytopenia with superadded infection usually implies a poor prognosis. Anemia can be due to a number of factors and does not necessarily imply bone marrow infiltration.
- Lymph node: Enlargement of the liver in 20%, spleen in 30% and lymph nodes in 50% of histiocytosis cases.
- Endocrine glands: Hypothalamic pituitary axisHypothalamic-pituitary-adrenal axisThe hypothalamic-pituitary-adrenal axis , also known as thelimbic-hypothalamic-pituitary-adrenal axis and, occasionally, as the hypothalamic-pituitary-adrenal-gonadotropic axis, is a complex set of direct influences and feedback interactions among the hypothalamus, the pituitary gland ,...
commonly involved. Diabetes insipidusDiabetes insipidusDiabetes insipidus is a condition characterized by excessive thirst and excretion of large amounts of severely diluted urine, with reduction of fluid intake having no effect on the concentration of the urine. There are several different types of DI, each with a different cause...
is most common. Anterior pituitaryAnterior pituitaryA major organ of the endocrine system, the anterior pituitary, also called the adenohypophysis, is the glandular, anterior lobe of the pituitary gland...
hormoneHormoneA hormone is a chemical released by a cell or a gland in one part of the body that sends out messages that affect cells in other parts of the organism. Only a small amount of hormone is required to alter cell metabolism. In essence, it is a chemical messenger that transports a signal from one...
deficiency is usually permanent. - Lungs: some patients are asymptomatic, diagnosed incidentally because of lung nodules on radiographs; others suffer from chronic cough and shortness of breath.
- Less frequently gastrointestinal tractGastrointestinal tractThe human gastrointestinal tract refers to the stomach and intestine, and sometimes to all the structures from the mouth to the anus. ....
and central nervous systemCentral nervous systemThe central nervous system is the part of the nervous system that integrates the information that it receives from, and coordinates the activity of, all parts of the bodies of bilaterian animals—that is, all multicellular animals except sponges and radially symmetric animals such as jellyfish...
.
Diagnosis
Diagnosis is confirmed histologically by tissueTissue (biology)
Tissue is a cellular organizational level intermediate between cells and a complete organism. A tissue is an ensemble of cells, not necessarily identical, but from the same origin, that together carry out a specific function. These are called tissues because of their identical functioning...
biopsy
Biopsy
A biopsy is a medical test involving sampling of cells or tissues for examination. It is the medical removal of tissue from a living subject to determine the presence or extent of a disease. The tissue is generally examined under a microscope by a pathologist, and can also be analyzed chemically...
. Hematoxylin-eosin stain of biopsy slide will show features of Langerhans cell e.g. distinct cell margin, pink granular cytoplasm
Cytoplasm
The cytoplasm is a small gel-like substance residing between the cell membrane holding all the cell's internal sub-structures , except for the nucleus. All the contents of the cells of prokaryote organisms are contained within the cytoplasm...
. Presence of Birbeck granules
Birbeck granules
Birbeck granules, also known as Birbeck bodies, are rod shaped or "tennis-racket" cytoplasmic organelles with a central linear density and a striated appearance...
on electron microscopy and immuno-cytochemical
Immunocytochemistry
Immunocytochemistry is a common laboratory technique that uses antibodies that target specific peptides or protein antigens in the cell via specific epitopes. These bound antibodies can then be detected using several different methods. ICC allows researchers to evaluate whether or not cells in a...
features e. g. CD1
CD1
For the album by Throbbing Gristle, see CD1 CD1 is a family of glycoproteins expressed on the surface of various human antigen-presenting cells. They are related to the class I MHC molecules, and are involved in the presentation of lipid antigens to T cells...
positivity are more specific. Initially routine blood tests e.g. full blood count, liver function test, U&Es, bone profile are done to determine disease extent and rule out other causes. Radiology will show osteolytic bone lesions and damage to the lung. The latter may be evident in chest X-ray
Chest X-ray
In medicine, a chest radiograph, commonly called a chest X-ray , is a projection radiograph of the chest used to diagnose conditions affecting the chest, its contents, and nearby structures...
s with micronodular and interstitial
Interstitial
An interstitial space or interstice is an empty space or gap between spaces full of structure or matter.In particular, interstitial may refer to:-Physical sciences:...
infiltrate in the mid and lower zone of lung, with sparing of the Costophrenic angle
Costophrenic angle
In anatomy, the costophrenic angles are the places where the diaphragm meet the ribs .Each costophrenic angle can normally be seen as on chest x-ray as a sharply-pointed, downward indentation between each hemi-diaphragm and the adjacent chest wall . A small portion of each lung normally reaches...
or honeycomb
Honeycomb
A honeycomb is a mass of hexagonal waxcells built by honey bees in their nests to contain their larvae and stores of honey and pollen.Beekeepers may remove the entire honeycomb to harvest honey...
appearance in older lesions. MRI and CT
Computed tomography
X-ray computed tomography or Computer tomography , is a medical imaging method employing tomography created by computer processing...
may show infiltration in sella turcica
Sella turcica
-External links:*...
. Assessment of endocrine function and bonemarrow biopsy are also performed when indicated.
Treatment
Treatment is guided by extent of disease. Solitary bone lesion may be amenable through excision or limited radiation. However systemic diseases often require chemotherapy. Use of systemic steroid is common, singly or adjunct to chemotherapy. Local steroid cream is applied to skin lesions. Endocrine deficiency often require lifelong supplement e.g. desmopressinDesmopressin
Desmopressin is a synthetic replacement for vasopressin, the hormone that reduces urine production. It may be taken nasally, intravenously, or as a tablet...
for diabetes insipidus which can be applied as nasal drop. Chemotherapeutic agents such as alkylating agents
Alkylating antineoplastic agent
An alkylating antineoplastic agent is an alkylating agent used in cancer treatment that attaches an alkyl group to DNA.The alkyl group is attached to the guanine base of DNA, at the number 7 nitrogen atom of the purine ring....
, antimetabolites, vinca alkaloids
Vinca alkaloids
Vinca alkaloids are a set of anti-mitotic and anti-microtubule agents which were originally derived from the Periwinkle plant Catharanthus roseus.Vinca alkaloids are used in the treatment of cancer...
either singly or in combination can lead to complete remission in diffuse disease.
Prognosis
Excellent for single foci disease. With multi-focal disease 60% have a chronic course, 30% achieve remission and mortality is up to 10%.External links
- http://www.eurohistio.net/index_eng.html a EU network granted by the EU commission with a multilanguage web site
- Images of LCH MedPix(r)Database
- Histiocyte Society An International Community Dedicated to Research and Treatment
- Seven Part Video Series
- Langerhans cell histiocytosis, article by the Sydney Children's Hospital
- 5-Minute Clinical Consult