
Cystic fibrosis transmembrane conductance regulator
Encyclopedia
Cystic fibrosis transmembrane conductance regulator (CFTR) is a protein
that in humans is encoded by the CFTR gene
.
CFTR is a ABC transporter-class ion channel
that transports chloride
and thiocyanate
ions across epithelial cell membrane
s. Mutations of the CFTR gene affect functioning of the chloride ion channels in these cell membranes, leading to cystic fibrosis
and congenital absence of the vas deferens
.
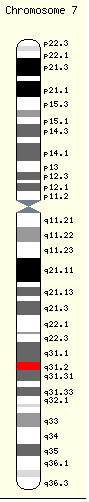 The gene that encodes the CFTR protein is found on the human
The gene that encodes the CFTR protein is found on the human
chromosome 7
, on the long arm at position q31.2. from base pair
116,907,253 to base pair 117,095,955. CFTR orthologs have also been identified in all mammals for which complete genome data are available.
The CFTR gene has been used in animals as a nuclear DNA
phylogenetic marker. Large genomic sequences of this gene have been used to explore the phylogeny of the major groups of mammals, and confirmed the grouping of placental orders into four major clades: Xenarthra
, Afrotheria
, Laurasiatheria
, and Euarchonta
plus Glires
.
s have been described that can affect the CFTR gene. Such mutations can cause two genetic disorders, congenital bilateral absence of vas deferens and the more widely known disorder cystic fibrosis
. Both disorders arise from the blockage of the movement of ions and, therefore, water into and out of cells. In congenital bilateral absence of vas deferens, the protein may be still functional but not at normal efficiency, this leads to the production of thick mucus
, which blocks the developing vas deferens
. In people with mutations giving rise to cystic fibrosis, the blockage in ion transport occurs in epithelial cells that line the passageways of the lungs, pancreas
, and other organs. This leads to chronic dysfunction, disability, and a reduced life expectancy.
The most common mutation, ΔF508 results from a deletion (Δ) of three nucleotides which results in a loss of the amino acid phenylalanine
(F) at the 508th position on the protein. As a result the protein does not fold
normally and is more quickly degraded.
The vast majority of mutations are quite rare. The distribution and frequency of mutations varies among different populations which has implications for genetic screening and counseling.
Mutations consist of replacements, duplications, deletions or shortenings in the CFTR gene. This may result in proteins that may not function, work less effectively, are more quickly degraded, or are present in inadequate numbers.
It has been hypothesized that mutations in the CFTR gene may confer a selective advantage to heterozygous individuals. Cells expressing a mutant form of the CFTR protein are resistant to invasion by the Salmonella typhi bacterium, the agent of typhoid fever, and mice carrying a single copy of mutant CFTR are resistant to diarrhea caused by cholera toxin.
 The most common mutations among caucasians
The most common mutations among caucasians
are:
with 1480 amino acid
s. The protein consists of five domains. There are two transmembrane domains, each with six spans of alpha helices
. These are each connected to a nucleotide binding domain (NBD) in the cytoplasm. The first NBD is connected to the second transmembrane domain by a regulatory "R" domain that is a unique feature of CFTR, not present in other ABC transporters. The ion channel only opens when its R-domain has been phosphorylated by PKA and ATP
is bound at the NBDs. The carboxyl terminal
of the protein is anchored to the cytoskeleton
by a PDZ-interacting domain.
-activated ATP
-gated anion channel
, increasing the conductance
for certain anions (e.g. Cl–) to flow down their electrochemical gradient
. ATP-driven conformational change
s, which in other ABC proteins fuel uphill substrate transport across cellular membranes, in CFTR open and close a gate to allow transmembrane flow of anions down their electrochemical gradient
. "Single CFTR channels open and close stochastically in an ATP
-dependent manner, the open state catalyzing exclusively "downhill" Cl– movement at rates of millions of ions per second, orders of magnitude too high for any enzymatic pump cycle to support." Essentially, CFTR is an ion channel that evolved as a 'broken' ABC transporter that leaks when in open conformation
.
The CFTR is found in the epithelial cells of many organs including the lung
, liver
, pancreas
, digestive
tract, reproductive tract, and skin
. Normally, the protein moves chloride
and thiocyanate
ion
s (with a negative charge) out of an epithelial cell to the covering mucus
. This results in an electrical gradient being formed and in the movement of (positively charged) sodium ions in the same direction as the chloride via a paracellular pathway. Due to this movement, the water potential of the mucus is reduced. This results in the movement of water out of the cell by osmosis
, and therefore a more fluid mucus.
In sweat gland
s, CFTR defects result in reduced transport of sodium chloride and sodium thiocyanate
in the reabsorptive duct and saltier sweat. This was the basis of a clinically important sweat test
for cystic fibrosis
before genetic screening was available.
with:
Protein
Proteins are biochemical compounds consisting of one or more polypeptides typically folded into a globular or fibrous form, facilitating a biological function. A polypeptide is a single linear polymer chain of amino acids bonded together by peptide bonds between the carboxyl and amino groups of...
that in humans is encoded by the CFTR gene
Gene
A gene is a molecular unit of heredity of a living organism. It is a name given to some stretches of DNA and RNA that code for a type of protein or for an RNA chain that has a function in the organism. Living beings depend on genes, as they specify all proteins and functional RNA chains...
.
CFTR is a ABC transporter-class ion channel
Ion channel
Ion channels are pore-forming proteins that help establish and control the small voltage gradient across the plasma membrane of cells by allowing the flow of ions down their electrochemical gradient. They are present in the membranes that surround all biological cells...
that transports chloride
Chloride
The chloride ion is formed when the element chlorine, a halogen, picks up one electron to form an anion Cl−. The salts of hydrochloric acid HCl contain chloride ions and can also be called chlorides. The chloride ion, and its salts such as sodium chloride, are very soluble in water...
and thiocyanate
Thiocyanate
Thiocyanate is the anion [SCN]−. It is the conjugate base of thiocyanic acid. Common derivatives include the colourless salts potassium thiocyanate and sodium thiocyanate. Organic compounds containing the functional group SCN are also called thiocyanates...
ions across epithelial cell membrane
Cell membrane
The cell membrane or plasma membrane is a biological membrane that separates the interior of all cells from the outside environment. The cell membrane is selectively permeable to ions and organic molecules and controls the movement of substances in and out of cells. It basically protects the cell...
s. Mutations of the CFTR gene affect functioning of the chloride ion channels in these cell membranes, leading to cystic fibrosis
Cystic fibrosis
Cystic fibrosis is a recessive genetic disease affecting most critically the lungs, and also the pancreas, liver, and intestine...
and congenital absence of the vas deferens
Congenital absence of the vas deferens
Congenital absence of the vas deferens is a condition in which the vasa deferentia, male reproductive organs, fail to form properly prior to birth. It may either be unilateral or bilateral .-Presentation:...
.
Gene
Human
Humans are the only living species in the Homo genus...
chromosome 7
Chromosome 7 (human)
Chromosome 7 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 7 spans more than 158 million base pairs and represents between 5 and 5.5 percent of the total DNA in cells.Identifying genes on each chromosome is an active area of genetic...
, on the long arm at position q31.2. from base pair
Base pair
In molecular biology and genetics, the linking between two nitrogenous bases on opposite complementary DNA or certain types of RNA strands that are connected via hydrogen bonds is called a base pair...
116,907,253 to base pair 117,095,955. CFTR orthologs have also been identified in all mammals for which complete genome data are available.
The CFTR gene has been used in animals as a nuclear DNA
Nuclear DNA
Nuclear DNA, nuclear deoxyribonucleic acid , is DNA contained within a nucleus of eukaryotic organisms. In mammals and vertebrates, nuclear DNA encodes more of the genome than the mitochondrial DNA and is composed of information inherited from two parents, one male, and one female, rather than...
phylogenetic marker. Large genomic sequences of this gene have been used to explore the phylogeny of the major groups of mammals, and confirmed the grouping of placental orders into four major clades: Xenarthra
Xenarthra
The superorder Xenarthra is a group of placental mammals , existent today only in the Americas and represented by anteaters, tree sloths, and armadillos. The origins of the order can be traced back as far as the Paleogene in South America...
, Afrotheria
Afrotheria
Afrotheria is a clade of mammals, the living members of which belong to groups from Africa or of African origin: golden moles, sengis , tenrecs, aardvarks, hyraxes, elephants and sea cows. The common ancestry of these animals was not recognized until the late 1990s...
, Laurasiatheria
Laurasiatheria
Laurasiatheria is a large group of placental mammals believed to have originated on the northern supercontinent of Laurasia. It includes shrews, hedgehogs, pangolins, bats, whales, most hoofed mammals, and carnivorans, among others....
, and Euarchonta
Euarchonta
The Euarchonta are a grandorder of mammals containing four orders: the Dermoptera or colugos, the Scandentia or treeshrews, the extinct Plesiadapiformes, and the Primates....
plus Glires
Glires
Glires is a clade consisting of rodents and lagomorphs . This hypothesis that these form a monophyletic group has been long debated based on morphological evidence, although recent morphological studies strongly support monophyly of Glires...
.
Mutations
Well over one thousand mutationMutation
In molecular biology and genetics, mutations are changes in a genomic sequence: the DNA sequence of a cell's genome or the DNA or RNA sequence of a virus. They can be defined as sudden and spontaneous changes in the cell. Mutations are caused by radiation, viruses, transposons and mutagenic...
s have been described that can affect the CFTR gene. Such mutations can cause two genetic disorders, congenital bilateral absence of vas deferens and the more widely known disorder cystic fibrosis
Cystic fibrosis
Cystic fibrosis is a recessive genetic disease affecting most critically the lungs, and also the pancreas, liver, and intestine...
. Both disorders arise from the blockage of the movement of ions and, therefore, water into and out of cells. In congenital bilateral absence of vas deferens, the protein may be still functional but not at normal efficiency, this leads to the production of thick mucus
Mucus
In vertebrates, mucus is a slippery secretion produced by, and covering, mucous membranes. Mucous fluid is typically produced from mucous cells found in mucous glands. Mucous cells secrete products that are rich in glycoproteins and water. Mucous fluid may also originate from mixed glands, which...
, which blocks the developing vas deferens
Vas deferens
The vas deferens , also called ductus deferens, , is part of the male anatomy of many vertebrates; they transport sperm from the epididymis in anticipation of ejaculation....
. In people with mutations giving rise to cystic fibrosis, the blockage in ion transport occurs in epithelial cells that line the passageways of the lungs, pancreas
Pancreas
The pancreas is a gland organ in the digestive and endocrine system of vertebrates. It is both an endocrine gland producing several important hormones, including insulin, glucagon, and somatostatin, as well as a digestive organ, secreting pancreatic juice containing digestive enzymes that assist...
, and other organs. This leads to chronic dysfunction, disability, and a reduced life expectancy.
The most common mutation, ΔF508 results from a deletion (Δ) of three nucleotides which results in a loss of the amino acid phenylalanine
Phenylalanine
Phenylalanine is an α-amino acid with the formula C6H5CH2CHCOOH. This essential amino acid is classified as nonpolar because of the hydrophobic nature of the benzyl side chain. L-Phenylalanine is an electrically neutral amino acid, one of the twenty common amino acids used to biochemically form...
(F) at the 508th position on the protein. As a result the protein does not fold
Protein folding
Protein folding is the process by which a protein structure assumes its functional shape or conformation. It is the physical process by which a polypeptide folds into its characteristic and functional three-dimensional structure from random coil....
normally and is more quickly degraded.
The vast majority of mutations are quite rare. The distribution and frequency of mutations varies among different populations which has implications for genetic screening and counseling.
Mutations consist of replacements, duplications, deletions or shortenings in the CFTR gene. This may result in proteins that may not function, work less effectively, are more quickly degraded, or are present in inadequate numbers.
It has been hypothesized that mutations in the CFTR gene may confer a selective advantage to heterozygous individuals. Cells expressing a mutant form of the CFTR protein are resistant to invasion by the Salmonella typhi bacterium, the agent of typhoid fever, and mice carrying a single copy of mutant CFTR are resistant to diarrhea caused by cholera toxin.
List of common mutations
Caucasian race
The term Caucasian race has been used to denote the general physical type of some or all of the populations of Europe, North Africa, the Horn of Africa, Western Asia , Central Asia and South Asia...
are:
- ΔF508
- G542X
- G551D
- N1303K
- W1282X
Structure
The CFTR gene is approximately 189 kb in length. CFTR is a glycoproteinGlycoprotein
Glycoproteins are proteins that contain oligosaccharide chains covalently attached to polypeptide side-chains. The carbohydrate is attached to the protein in a cotranslational or posttranslational modification. This process is known as glycosylation. In proteins that have segments extending...
with 1480 amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
s. The protein consists of five domains. There are two transmembrane domains, each with six spans of alpha helices
Alpha helix
A common motif in the secondary structure of proteins, the alpha helix is a right-handed coiled or spiral conformation, in which every backbone N-H group donates a hydrogen bond to the backbone C=O group of the amino acid four residues earlier...
. These are each connected to a nucleotide binding domain (NBD) in the cytoplasm. The first NBD is connected to the second transmembrane domain by a regulatory "R" domain that is a unique feature of CFTR, not present in other ABC transporters. The ion channel only opens when its R-domain has been phosphorylated by PKA and ATP
Adenosine triphosphate
Adenosine-5'-triphosphate is a multifunctional nucleoside triphosphate used in cells as a coenzyme. It is often called the "molecular unit of currency" of intracellular energy transfer. ATP transports chemical energy within cells for metabolism...
is bound at the NBDs. The carboxyl terminal
C-terminal end
The C-terminus is the end of an amino acid chain , terminated by a free carboxyl group . When the protein is translated from messenger RNA, it is created from N-terminus to C-terminus...
of the protein is anchored to the cytoskeleton
Cytoskeleton
The cytoskeleton is a cellular "scaffolding" or "skeleton" contained within a cell's cytoplasm and is made out of protein. The cytoskeleton is present in all cells; it was once thought to be unique to eukaryotes, but recent research has identified the prokaryotic cytoskeleton...
by a PDZ-interacting domain.
Function
CFTR functions as a cAMPCyclic adenosine monophosphate
Cyclic adenosine monophosphate is a second messenger important in many biological processes...
-activated ATP
Adenosine triphosphate
Adenosine-5'-triphosphate is a multifunctional nucleoside triphosphate used in cells as a coenzyme. It is often called the "molecular unit of currency" of intracellular energy transfer. ATP transports chemical energy within cells for metabolism...
-gated anion channel
Ion channel
Ion channels are pore-forming proteins that help establish and control the small voltage gradient across the plasma membrane of cells by allowing the flow of ions down their electrochemical gradient. They are present in the membranes that surround all biological cells...
, increasing the conductance
Conductance
Conductance may refer to:* Electrical conductance, the ability for electricity to flow a certain path* Fluid conductance, the ability for fluid to transmit through materials* Thermal conductivity, the ability for temperatures to transmit through materials...
for certain anions (e.g. Cl–) to flow down their electrochemical gradient
Electrochemical gradient
An electrochemical gradient is a spatial variation of both electrical potential and chemical concentration across a membrane; that is, a combination of the membrane potential and the pH gradient...
. ATP-driven conformational change
Conformational change
A macromolecule is usually flexible and dynamic. It can change its shape in response to changes in its environment or other factors; each possible shape is called a conformation, and a transition between them is called a conformational change...
s, which in other ABC proteins fuel uphill substrate transport across cellular membranes, in CFTR open and close a gate to allow transmembrane flow of anions down their electrochemical gradient
Electrochemical gradient
An electrochemical gradient is a spatial variation of both electrical potential and chemical concentration across a membrane; that is, a combination of the membrane potential and the pH gradient...
. "Single CFTR channels open and close stochastically in an ATP
Adenosine triphosphate
Adenosine-5'-triphosphate is a multifunctional nucleoside triphosphate used in cells as a coenzyme. It is often called the "molecular unit of currency" of intracellular energy transfer. ATP transports chemical energy within cells for metabolism...
-dependent manner, the open state catalyzing exclusively "downhill" Cl– movement at rates of millions of ions per second, orders of magnitude too high for any enzymatic pump cycle to support." Essentially, CFTR is an ion channel that evolved as a 'broken' ABC transporter that leaks when in open conformation
Chemical structure
A chemical structure includes molecular geometry, electronic structure and crystal structure of molecules. Molecular geometry refers to the spatial arrangement of atoms in a molecule and the chemical bonds that hold the atoms together. Molecular geometry can range from the very simple, such as...
.
The CFTR is found in the epithelial cells of many organs including the lung
Lung
The lung is the essential respiration organ in many air-breathing animals, including most tetrapods, a few fish and a few snails. In mammals and the more complex life forms, the two lungs are located near the backbone on either side of the heart...
, liver
Liver
The liver is a vital organ present in vertebrates and some other animals. It has a wide range of functions, including detoxification, protein synthesis, and production of biochemicals necessary for digestion...
, pancreas
Pancreas
The pancreas is a gland organ in the digestive and endocrine system of vertebrates. It is both an endocrine gland producing several important hormones, including insulin, glucagon, and somatostatin, as well as a digestive organ, secreting pancreatic juice containing digestive enzymes that assist...
, digestive
Digestive
Digestive may refer to:*Digestion, biological process of metabolism*Digestive biscuit, a British semi-sweet biscuit*Digestif, small beverage at the end of a meal...
tract, reproductive tract, and skin
Skin
-Dermis:The dermis is the layer of skin beneath the epidermis that consists of connective tissue and cushions the body from stress and strain. The dermis is tightly connected to the epidermis by a basement membrane. It also harbors many Mechanoreceptors that provide the sense of touch and heat...
. Normally, the protein moves chloride
Chloride
The chloride ion is formed when the element chlorine, a halogen, picks up one electron to form an anion Cl−. The salts of hydrochloric acid HCl contain chloride ions and can also be called chlorides. The chloride ion, and its salts such as sodium chloride, are very soluble in water...
and thiocyanate
Thiocyanate
Thiocyanate is the anion [SCN]−. It is the conjugate base of thiocyanic acid. Common derivatives include the colourless salts potassium thiocyanate and sodium thiocyanate. Organic compounds containing the functional group SCN are also called thiocyanates...
ion
Ion
An ion is an atom or molecule in which the total number of electrons is not equal to the total number of protons, giving it a net positive or negative electrical charge. The name was given by physicist Michael Faraday for the substances that allow a current to pass between electrodes in a...
s (with a negative charge) out of an epithelial cell to the covering mucus
Mucus
In vertebrates, mucus is a slippery secretion produced by, and covering, mucous membranes. Mucous fluid is typically produced from mucous cells found in mucous glands. Mucous cells secrete products that are rich in glycoproteins and water. Mucous fluid may also originate from mixed glands, which...
. This results in an electrical gradient being formed and in the movement of (positively charged) sodium ions in the same direction as the chloride via a paracellular pathway. Due to this movement, the water potential of the mucus is reduced. This results in the movement of water out of the cell by osmosis
Osmosis
Osmosis is the movement of solvent molecules through a selectively permeable membrane into a region of higher solute concentration, aiming to equalize the solute concentrations on the two sides...
, and therefore a more fluid mucus.
In sweat gland
Sweat gland
Sweat glands, or sudoriferous glands, are small tubular structures of the skin that produce sweat. There are two kinds of sweat glands:...
s, CFTR defects result in reduced transport of sodium chloride and sodium thiocyanate
Thiocyanate
Thiocyanate is the anion [SCN]−. It is the conjugate base of thiocyanic acid. Common derivatives include the colourless salts potassium thiocyanate and sodium thiocyanate. Organic compounds containing the functional group SCN are also called thiocyanates...
in the reabsorptive duct and saltier sweat. This was the basis of a clinically important sweat test
Sweat test
The sweat test measures the concentration of chloride that is excreted in sweat. It is used to screen for cystic fibrosis .-Background:Cystic fibrosis is caused by defects in a protein found in many tissues, including the airways and the sweat glands. As a result, these tissues do not work properly...
for cystic fibrosis
Cystic fibrosis
Cystic fibrosis is a recessive genetic disease affecting most critically the lungs, and also the pancreas, liver, and intestine...
before genetic screening was available.
Interactions
Cystic fibrosis transmembrane conductance regulator has been shown to interactProtein-protein interaction
Protein–protein interactions occur when two or more proteins bind together, often to carry out their biological function. Many of the most important molecular processes in the cell such as DNA replication are carried out by large molecular machines that are built from a large number of protein...
with:
- DNAJC5DNAJC5DnaJ homolog subfamily C member 5, also known as cysteine string protein or CSP is a protein, that in humans encoded by the DNAJC5 gene. It was first described in 1990.- Gene :...
, - GOPCGOPCGolgi-associated PDZ and coiled-coil motif-containing protein is a protein that in humans is encoded by the GOPC gene.-Interactions:GOPC has been shown to interact with GRID2, BECN1, RHOQ, ACCN3, Cystic fibrosis transmembrane conductance regulator and CSPG5.-Further reading:...
, - PDZK1PDZK1Na/H exchange regulatory cofactor NHE-RF3 is a protein that in humans is encoded by the PDZK1 gene.-Interactions:PDZK1 has been shown to interact with AKAP10, FARP2, Sodium-hydrogen antiporter 3 regulator 1, SLC22A12, SLK, SLC22A4, CLCN3, PDZK1IP1, Cystic fibrosis transmembrane conductance...
, - PRKCEPRKCEProtein kinase C epsilon type is an enzyme that in humans is encoded by the PRKCE gene.- Function :Protein kinase C is a family of serine- and threonine-specific protein kinases that can be activated by calcium and the second messenger diacylglycerol...
, - SLC4A8SLC4A8Electroneutral sodium bicarbonate exchanger 1 is a protein that in humans is encoded by the SLC4A8 gene.-Interactions:SLC4A8 has been shown to interact with Sodium-hydrogen antiporter 3 regulator 1 and Cystic fibrosis transmembrane conductance regulator.-Further reading:...
, - SNAP23SNAP23Synaptosomal-associated protein 23 is a protein that in humans is encoded by the SNAP23 gene. Two alternative transcript variants encoding different protein isoforms have been described for this gene.- Function :...
, - SLC9A3R1Sodium-hydrogen antiporter 3 regulator 1Sodium-hydrogen antiporter 3 regulator 1 is a regulator of Sodium-hydrogen antiporter 3. It is encoded by the gene SLC9A3R1. It is also known as ERM Binding Protein 50 or Na+/H+ Exchanger Regulatory Factor...
, - SLC9A3R2Sodium-hydrogen exchange regulatory cofactor 2Sodium-hydrogen exchange regulatory cofactor NHE-RF2 also known as tyrosine kinase activator protein 1 or SRY-interacting protein 1 is a protein that in humans is encoded by the SLC9A3R2 gene.NHERF-2) is a scaffold protein that connects plasma membrane proteins with members of the...
, and - STX1ASTX1ASyntaxin-1A is a protein that in humans is encoded by the STX1A gene.- Function :Synaptic vesicles store neurotransmitters that are released during calcium-regulated exocytosis. The specificity of neurotransmitter release requires the localization of both synaptic vesicles and calcium channels to...
,
Related conditions
- Congenital bilateral absence of vas deferens: Males with congenital bilateral absence of the vas deferensVas deferensThe vas deferens , also called ductus deferens, , is part of the male anatomy of many vertebrates; they transport sperm from the epididymis in anticipation of ejaculation....
most often have a mild mutationMutationIn molecular biology and genetics, mutations are changes in a genomic sequence: the DNA sequence of a cell's genome or the DNA or RNA sequence of a virus. They can be defined as sudden and spontaneous changes in the cell. Mutations are caused by radiation, viruses, transposons and mutagenic...
(a change that allows partial function of the gene) in one copy of the CFTR gene and a cystic fibrosis-causing mutation in the other copy of CFTR. As a result of these mutations, the movement of water and salt into and out of cells is disrupted. This disturbance leads to the production of a large amount of thick mucus that blocks the developing vas deferens (a tube that carries sperm from the testes) and causes it to degenerate, resulting in infertility.
- Cystic fibrosisCystic fibrosisCystic fibrosis is a recessive genetic disease affecting most critically the lungs, and also the pancreas, liver, and intestine...
: More than 1,700 mutations in the CFTR gene have been found but the majority of these have not been associated with cystic fibrosis. Most of these mutations either substitute one amino acidAmino acidAmino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
(a building block of proteins) for another amino acid in the CFTR protein or delete a small amount of DNADNADeoxyribonucleic acid is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms . The DNA segments that carry this genetic information are called genes, but other DNA sequences have structural purposes, or are involved in...
in the CFTR gene. The most common mutation, called ΔF508, is a deletion (Δ) of one amino acid (phenylalanine) at position 508 in the CFTR protein. This altered protein never reaches the cell membrane because it is degraded shortly after it is made. All disease-causing mutations in the CFTR gene prevent the channel from functioning properly, leading to a blockage of the movement of salt and water into and out of cells. As a result of this blockage, cells that line the passageways of the lungs, pancreas, and other organs produce abnormally thick, sticky mucus. This mucus obstructs the airways and glands, causing the characteristic signs and symptoms of cystic fibrosis. In addition, only thin mucus can be removed by ciliaCiliumA cilium is an organelle found in eukaryotic cells. Cilia are slender protuberances that project from the much larger cell body....
, thick mucus cannot, so it traps bacteria that give rise to chronic infections.
External links
- GeneReviews/NCBI/NIH/UW entry on CFTR-Related Disorders - Cystic Fibrosis (CF, Mucoviscidosis) and Congenital Absence of the Vas Deferens (CAVD)
- The Cystic Fibrosis Transmembrane Conductance Regulator Protein
- The Human Gene Mutation Database - CFTR Records
- Cystic Fibrosis Mutation Database
- Oak Ridge National Laboratory CFTR Information
- CFTR at OMIM (National Center for Biotechnology Information)