

Niemann-Pick disease
Encyclopedia
Niemann–Pick disease refers to a group of fatal inherited metabolic disorders that are included in the larger family of lysosomal storage disease
s (LSDs).
) may cause reduced appetite, abdominal distension and pain as well as thrombocytopenia
secondary to splenomegaly.
Sphingomyelin
accumulation in the central nervous system
(including the cerebellum
) results in unsteady gait (ataxia
), slurring of speech (dysarthria
) and discoordinated swallowing (dysphagia
). Basal ganglia
dysfunction causes abnormal posturing of the limbs, trunk and face (dystonia
) and upper brainstem disease results in impaired voluntary rapid eye movements (supranuclear gaze palsy). More widespread disease involving the cerebral cortex
and subcortical structures is responsible for gradual loss of intellectual abilities causing dementia
and seizure
s.
Bone marrow
cavities may be enlarged and the cortical bone
may be thinned. Coxa vara
may be found.
Sleep related disorders are also seen, including gelastic cataplexy
(sudden loss of muscle tone associated with laughter), and sleep inversion (sleepiness during the day and wakefulness at night).
and over eating
 Mutations in the SMPD1 gene
Mutations in the SMPD1 gene
cause Niemann–Pick disease types A and B, and mutations in NPC1
and NPC2
cause Niemann-Pick disease, type C
(NPC). Type D was originally separated from Type C to delineate a group of patients with otherwise identical disorders who shared a common Nova Scotian ancestry. Patients in this group are now known to share a specific mutation in the NPC 1 gene, and NPC is now used to embrace both groups. The terms "Niemann-Pick type I" and "Niemann-Pick type II" were proposed to separate the high and low sphingomyelin forms of the disease in the early 1980s, before the molecular defects were described.
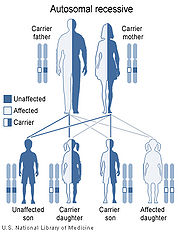
Niemann–Pick disease is inherited in an autosomal recessive pattern, which means both copies, or allele
s, of the gene must be mutated (altered in such a way that function is impaired, in contrast to a polymorphism
, in which the nucleotide
sequence is altered but causes no functional disruption) for a person to be affected by the disorder. Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene. If both parents are carriers, there is a 25% chance with each pregnancy for an affected child. Genetic counseling
and genetic testing
is recommended for families who may be carriers of Niemann-Pick.
In April 2009, a promising development in treatment of Niemann Pick type C was announced. The U.S. Food and Drug Administration approved Investigational New Drug (INDs) applications for six year old identical twins, Addison and Cassidy Hempel of Reno, Nevada, to receive intravenous infusions of hydroxy-propyl-β-cyclodextrin
or HPBCD. The twins are the first in the United States to receive experimental treatment with HPBCD and are currently undergoing treatment at Renown Regional Medical Center in Nevada.
In February 2010, the parents of the twins with the support of Children's Hospital Research Center Oakland, filed an orphan drug application with the Office of Orphan Product Development at the FDA for Trappsol brand cyclodextrin for the treatment of NPC disease. On May 17, 2010, the FDA granted hydroxy-propyl-beta-cyclodextrin orphan drug status and designated hydroxy-propyl-beta-cyclodextrin as a treatment for Niemann Pick Type C disease.
On July 14, 2010, Children's Hospital Research Center Oakland filed new Investigational New Drug (INDs) applications with the FDA on behalf of the Hempel twins based on promising new animal data. Doctors have requested to deliver hydroxy-propyl-beta-cyclodextrin intrathecally and directly into the central nervous system of the twins in an attempt to arrest neurodegeneration. The INDs were approved on September 23, 2010, and treatments are expected to start on October 15, 2010.
published the first description of what is now known as Niemann–Pick disease, type A, in 1914. Ludwig Pick
described the pathology of the disease in a series of papers in the 1930s.
Lysosomal storage disease
Lysosomal storage diseases are a group of approximately 50 rare inherited metabolic disorders that result from defects in lysosomal function...
s (LSDs).
Signs and symptoms
Symptoms are related to the organs in which they accumulate. Enlargement of the liver and spleen (hepatosplenomegalyHepatosplenomegaly
Hepatosplenomegaly is the simultaneous enlargement of both the liver and the spleen . Hepatosplenomegaly can occur as the result of acute viral hepatitis or infectious mononucleosis, or it can be the sign of a serious and life threatening lysosomal storage disease...
) may cause reduced appetite, abdominal distension and pain as well as thrombocytopenia
Thrombocytopenia
Thrombocytopenia is a relative decrease of platelets in blood.A normal human platelet count ranges from 150,000 to 450,000 platelets per microliter of blood. These limits are determined by the 2.5th lower and upper percentile, so values outside this range do not necessarily indicate disease...
secondary to splenomegaly.
Sphingomyelin
Sphingomyelin
Sphingomyelin is a type of sphingolipid found in animal cell membranes, especially in the membranous myelin sheath that surrounds some nerve cell axons. It usually consists of phosphorylcholine and ceramide...
accumulation in the central nervous system
Central nervous system
The central nervous system is the part of the nervous system that integrates the information that it receives from, and coordinates the activity of, all parts of the bodies of bilaterian animals—that is, all multicellular animals except sponges and radially symmetric animals such as jellyfish...
(including the cerebellum
Cerebellum
The cerebellum is a region of the brain that plays an important role in motor control. It may also be involved in some cognitive functions such as attention and language, and in regulating fear and pleasure responses, but its movement-related functions are the most solidly established...
) results in unsteady gait (ataxia
Ataxia
Ataxia is a neurological sign and symptom that consists of gross lack of coordination of muscle movements. Ataxia is a non-specific clinical manifestation implying dysfunction of the parts of the nervous system that coordinate movement, such as the cerebellum...
), slurring of speech (dysarthria
Dysarthria
Dysarthria is a motor speech disorder resulting from neurological injury of the motor component of the motor-speech system and is characterized by poor articulation of phonemes...
) and discoordinated swallowing (dysphagia
Dysphagia
Dysphagia is the medical term for the symptom of difficulty in swallowing. Although classified under "symptoms and signs" in ICD-10, the term is sometimes used as a condition in its own right. Sufferers are sometimes unaware of their dysphagia....
). Basal ganglia
Basal ganglia
The basal ganglia are a group of nuclei of varied origin in the brains of vertebrates that act as a cohesive functional unit. They are situated at the base of the forebrain and are strongly connected with the cerebral cortex, thalamus and other brain areas...
dysfunction causes abnormal posturing of the limbs, trunk and face (dystonia
Dystonia
Dystonia is a neurological movement disorder, in which sustained muscle contractions cause twisting and repetitive movements or abnormal postures. The disorder may be hereditary or caused by other factors such as birth-related or other physical trauma, infection, poisoning or reaction to...
) and upper brainstem disease results in impaired voluntary rapid eye movements (supranuclear gaze palsy). More widespread disease involving the cerebral cortex
Cerebral cortex
The cerebral cortex is a sheet of neural tissue that is outermost to the cerebrum of the mammalian brain. It plays a key role in memory, attention, perceptual awareness, thought, language, and consciousness. It is constituted of up to six horizontal layers, each of which has a different...
and subcortical structures is responsible for gradual loss of intellectual abilities causing dementia
Dementia
Dementia is a serious loss of cognitive ability in a previously unimpaired person, beyond what might be expected from normal aging...
and seizure
Seizure
An epileptic seizure, occasionally referred to as a fit, is defined as a transient symptom of "abnormal excessive or synchronous neuronal activity in the brain". The outward effect can be as dramatic as a wild thrashing movement or as mild as a brief loss of awareness...
s.
Bone marrow
Bone marrow
Bone marrow is the flexible tissue found in the interior of bones. In humans, bone marrow in large bones produces new blood cells. On average, bone marrow constitutes 4% of the total body mass of humans; in adults weighing 65 kg , bone marrow accounts for approximately 2.6 kg...
cavities may be enlarged and the cortical bone
Cortical bone
Cortical bone, synonymous with compact bone, is one of the two types of osseous tissue that form bones. Cortical bone facilitates bone's main functions: to support the whole body, protect organs, provide levers for movement, and store and release chemical elements, mainly calcium. As its name...
may be thinned. Coxa vara
Coxa vara
Coxa vara is a deformity of the hip, whereby the angle between the ball and the shaft of the femur is reduced to less than 120 degrees. This results in the leg being shortened, and therefore a limp occurs. It is commonly caused by injury, such as a fracture...
may be found.
Sleep related disorders are also seen, including gelastic cataplexy
Cataplexy
Cataplexy is a sudden and transient episode of loss of muscle tone, often triggered by emotions. It is a rare disease , but affects roughly 70% of people who have narcolepsy...
(sudden loss of muscle tone associated with laughter), and sleep inversion (sleepiness during the day and wakefulness at night).
and over eating
Cause and genetics
Gene
A gene is a molecular unit of heredity of a living organism. It is a name given to some stretches of DNA and RNA that code for a type of protein or for an RNA chain that has a function in the organism. Living beings depend on genes, as they specify all proteins and functional RNA chains...
cause Niemann–Pick disease types A and B, and mutations in NPC1
NPC1
Niemann-Pick disease, type C1 also known as NPC1 is a protein which in humans is encoded by the NPC1 gene.NPC1 was identified as the gene that when mutated, results in Niemann-Pick disease, type C...
and NPC2
NPC2
NPC2 is a protein associated with Niemann-Pick disease, type C....
cause Niemann-Pick disease, type C
Niemann-Pick disease, type C
Niemann-Pick type C is a lysosomal storage disease associated with mutations in NPC1 and NPC2 genes. Niemann-Pick Type C strikes an estimated 1:150,000 people...
(NPC). Type D was originally separated from Type C to delineate a group of patients with otherwise identical disorders who shared a common Nova Scotian ancestry. Patients in this group are now known to share a specific mutation in the NPC 1 gene, and NPC is now used to embrace both groups. The terms "Niemann-Pick type I" and "Niemann-Pick type II" were proposed to separate the high and low sphingomyelin forms of the disease in the early 1980s, before the molecular defects were described.
Niemann–Pick disease is inherited in an autosomal recessive pattern, which means both copies, or allele
Allele
An allele is one of two or more forms of a gene or a genetic locus . "Allel" is an abbreviation of allelomorph. Sometimes, different alleles can result in different observable phenotypic traits, such as different pigmentation...
s, of the gene must be mutated (altered in such a way that function is impaired, in contrast to a polymorphism
Polymorphism (biology)
Polymorphism in biology occurs when two or more clearly different phenotypes exist in the same population of a species — in other words, the occurrence of more than one form or morph...
, in which the nucleotide
Nucleotide
Nucleotides are molecules that, when joined together, make up the structural units of RNA and DNA. In addition, nucleotides participate in cellular signaling , and are incorporated into important cofactors of enzymatic reactions...
sequence is altered but causes no functional disruption) for a person to be affected by the disorder. Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene. If both parents are carriers, there is a 25% chance with each pregnancy for an affected child. Genetic counseling
Genetic counseling
Genetic counseling or traveling is the process by which patients or relatives, at risk of an inherited disorder, are advised of the consequences and nature of the disorder, the probability of developing or transmitting it, and the options open to them in management and family planning...
and genetic testing
Genetic testing
Genetic testing is among the newest and most sophisticated of techniques used to test for genetic disorders which involves direct examination of the DNA molecule itself. Other genetic tests include biochemical tests for such gene products as enzymes and other proteins and for microscopic...
is recommended for families who may be carriers of Niemann-Pick.
Classification
In 1961, the following classification was introduced:In April 2009, a promising development in treatment of Niemann Pick type C was announced. The U.S. Food and Drug Administration approved Investigational New Drug (INDs) applications for six year old identical twins, Addison and Cassidy Hempel of Reno, Nevada, to receive intravenous infusions of hydroxy-propyl-β-cyclodextrin
Cyclodextrin
Cyclodextrins are a family of compounds made up of sugar molecules bound together in a ring ....
or HPBCD. The twins are the first in the United States to receive experimental treatment with HPBCD and are currently undergoing treatment at Renown Regional Medical Center in Nevada.
In February 2010, the parents of the twins with the support of Children's Hospital Research Center Oakland, filed an orphan drug application with the Office of Orphan Product Development at the FDA for Trappsol brand cyclodextrin for the treatment of NPC disease. On May 17, 2010, the FDA granted hydroxy-propyl-beta-cyclodextrin orphan drug status and designated hydroxy-propyl-beta-cyclodextrin as a treatment for Niemann Pick Type C disease.
On July 14, 2010, Children's Hospital Research Center Oakland filed new Investigational New Drug (INDs) applications with the FDA on behalf of the Hempel twins based on promising new animal data. Doctors have requested to deliver hydroxy-propyl-beta-cyclodextrin intrathecally and directly into the central nervous system of the twins in an attempt to arrest neurodegeneration. The INDs were approved on September 23, 2010, and treatments are expected to start on October 15, 2010.
Prognosis
Type A Niemann Pick disease has an extremely poor prognosis with most cases being fatal by the age of 18 months. Type B and C Niemann Pick disease have a better prognosis, with many patients with these disorders living into their teens orHistory
Albert NiemannAlbert Niemann (paediatrician)
Albert Niemann was a German physician.Niemann-Pick disease is named for him and Ludwig Pick.He was the son of Albert Niemann, a well known tenor with the same name.-References:...
published the first description of what is now known as Niemann–Pick disease, type A, in 1914. Ludwig Pick
Ludwig Pick
Professor Ludwig Pick was a German pathologist who was a native of Landsberg an der Warthe. In 1893 he earned his medical doctorate in Leipzig, and subsequently practiced medicine at Leopold Landau's private Frauenklinik, where he remained until 1906...
described the pathology of the disease in a series of papers in the 1930s.
External links
- National Niemann-Pick Disease Foundation (U.S.)
- Ara Parseghian Medical Research Foundation
- Niemann-Pick Disease Group (UK)
- GeneReviews/NCBI/NIH/UW entry on Acid Sphingomyelinase Deficiency Includes: Niemann-Pick Disease Type A, Niemann-Pick Disease Type B
- OMIM entries on Acid Sphingomyelinase Deficiency
- GeneReviews/NCBI/NIH/UW entry on Niemann-Pick Disease Type C
- OMIM entries on Niemann-Pick Type C
- Niemann-Pick Disease Group Canada
- National Institutes of Health Clinical Center Study On Niemann Pick Type C
- Marc C. Patterson, MD, child neurologist, Mayo Clinic
- National Institute of Neurological Diseaases and Strokes
- Addi and Cassi Hempel: Identical twins with Niemann Pick type C disease being treated with cyclodextrin
- Genetics Home Reference on Niemann Pick Disease
- Hide & Seek Foundation for Lysosomal Disease Research
- Detailed information about Niemann Pick Type C for patients and Healthcare Professionals
- Pathology images and digital slides (HP:7983)
- This article incorporates public domain text from The U.S. National Library of Medicine