

Zero-differential overlap
Encyclopedia
Zero differential overlap is an approximation that is used to ignore certain integrals, usually two-electron repulsion integrals, in semi-empirical quantum chemistry methods
quantum chemistry
molecular orbital
methods.
If the molecular orbitals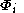 are expanded in terms of N basis functions,
are expanded in terms of N basis functions, 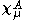 as:-
as:-
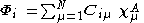
where A is the atom the basis function is centred on, and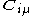 are coefficients, the two-electron repulsion integrals are then defined as:-
are coefficients, the two-electron repulsion integrals are then defined as:-
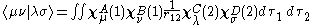
The zero differential overlap approximation ignores integrals that contain the product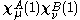 where μ is not equal to ν. This leads to:-
where μ is not equal to ν. This leads to:-
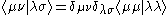
where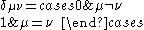
The total number of such integrals is reduced to N(N + 1) / 2 (approximately N2 / 2) from [N(N + 1) / 2][N(N + 1) / 2 + 1] / 2 (approximately N4 / 8), all of which are included in ab initio
Hartree–Fock and post-Hartree–Fock calculations.
Methods such as the Pariser–Parr–Pople method (PPP) and CNDO/2
use the zero differential overlap approximation completely. Methods based on the intermediate neglect of differential overlap, such as INDO
, MINDO
, ZINDO
and SINDO
do not apply it when A = B = C = D, i.e. when all four basis functions are on the same atom. Methods that use the neglect of diatomic differential overlap, such as MNDO
, PM3
and AM1, also do not apply it when A = B and C = D, i.e. when the basis functions for the first electron are on the same atom and the basis functions for the second electron are the same atom.
It is possible to partly justify this approximation, but generally it is used because it works reasonably well when the integrals that remain –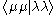 – are parameterised.
– are parameterised.
Semi-empirical quantum chemistry methods
Semi-empirical quantum chemistry methods are based on the Hartree-Fock formalism, but make many approximations and obtain some parameters from empirical data. They are very important in computational chemistry for treating large molecules where the full Hartree-Fock method without the...
quantum chemistry
Quantum chemistry
Quantum chemistry is a branch of chemistry whose primary focus is the application of quantum mechanics in physical models and experiments of chemical systems...
molecular orbital
Molecular orbital
In chemistry, a molecular orbital is a mathematical function describing the wave-like behavior of an electron in a molecule. This function can be used to calculate chemical and physical properties such as the probability of finding an electron in any specific region. The term "orbital" was first...
methods.
If the molecular orbitals
are expanded in terms of N basis functions, as:-where A is the atom the basis function is centred on, and
are coefficients, the two-electron repulsion integrals are then defined as:-The zero differential overlap approximation ignores integrals that contain the product
where μ is not equal to ν. This leads to:-where
The total number of such integrals is reduced to N(N + 1) / 2 (approximately N2 / 2) from [N(N + 1) / 2][N(N + 1) / 2 + 1] / 2 (approximately N4 / 8), all of which are included in ab initio
Ab initio quantum chemistry methods
Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initiowas first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited states of benzene.The background is described by Parr...
Hartree–Fock and post-Hartree–Fock calculations.
Methods such as the Pariser–Parr–Pople method (PPP) and CNDO/2
CNDO/2
CNDO is the abbreviation for Complete Neglect of Differential Overlap. Although CNDO is based on quantum chemistry, it is more specifically one of the first semi-empirical quantum chemistry methods. It uses two approximations:...
use the zero differential overlap approximation completely. Methods based on the intermediate neglect of differential overlap, such as INDO
INDO
INDO stands for Intermediate Neglect of Differential Overlap. It is a semi-empirical quantum chemistry method that is a development of the complete neglect of differential overlap method introduced by John Pople...
, MINDO
MINDO
MINDO, or Modified Intermediate Neglect of Differential Overlap is a semi-empirical method for the quantum calculation of molecular electronic structure in computational chemistry. It is based on the Intermediate Neglect of Differential Overlap method of John Pople. It was developed by the group...
, ZINDO
ZINDO
ZINDO is a semi-empirical quantum chemistry method used in computational chemistry. It is a development of the INDO method. It stands for Zerner's Intermediate Neglect of Differential Overlap, as it was developed by Michael Zerner...
and SINDO
SINDO
SINDO, or actually SINDO1, is one of many semi-empirical quantum chemistry methods. It stands for symmetric orthogonalised INDO and was developed by K. Jug and coworkers. Like MINDO, it is a development of the INDO method. The main development is the inclusion of d orbitals for atoms of the...
do not apply it when A = B = C = D, i.e. when all four basis functions are on the same atom. Methods that use the neglect of diatomic differential overlap, such as MNDO
MNDO
MNDO, or Modified Neglect of Differential Overlap is a semi-empirical method for the quantum calculation of molecular electronic structure in computational chemistry. It is based on the Neglect of Differential Diatomic Overlap integral approximation. Similarly, this method replaced the earlier...
, PM3
PM3 (chemistry)
PM3, or Parameterized Model number 3, is a semi-empirical method for the quantum calculation of molecular electronic structure in computational chemistry. It is based on the Neglect of Differential Diatomic Overlap integral approximation....
and AM1, also do not apply it when A = B and C = D, i.e. when the basis functions for the first electron are on the same atom and the basis functions for the second electron are the same atom.
It is possible to partly justify this approximation, but generally it is used because it works reasonably well when the integrals that remain –
– are parameterised.