
.gif)
Coot (program)
Encyclopedia
The program Coot is used to display and manipulate atomic models of macromolecules, typically of proteins or nucleic acids, using 3D computer graphics
. It is primary focussed on the building and validation of atomic models into 3-dimensional electron density
maps obtained by X-ray crystallography
methods, although it has also been applied to data from electron microscopy.
. The software is designed to be easy-to-learn for novice users, achieved by ensuring that tools for common tasks are 'discoverable' through familiar user interface elements (menus and toolbars), or by intuitive behaviour (mouse controls). Recent developments have enhanced the usability of the software for expert users, with customisable key bindings, extensions, and an extensive scripting interface.
Coot is free software
, distributed under the GNU GPL. It is available from the Coot web site at the University of York. Pre-compiled binaries are available for Linux and Windows from York, and for Mac OS X through Fink
. Additional support is available through the Coot wiki.
The primary author is Paul Emsley (University of Oxford). Other contributors include Kevin Cowtan, Bernhard Lohkamp and Stuart McNicholas (University of York) and William Scott and Eugene Krissinel.
, mmcif, and Shelx files. The model may then be rotated in 3D and viewed from any viewpoint. The atomic model is represented by default using a stick-model, with vectors representing chemical bonds. The two halves of each bond are coloured according to the element of the atom at that end of the bond, allowing chemical structure and identity to be visualised in a manner familiar to most chemists.
Coot can also display electron density, which is the result of structure determination experiments such as X-ray crystallography and EM reconstruction. The density is contoured using a 3D-mesh. The contour level controlled using the mouse wheel for easy manipulation - this provides a simple way for the user to get an idea of the 3D electron density profile without the visual clutter of multiple contour levels. Electron density may be read into the program from ccp4
or cns
map formats, how it is more normal to calculate an electron density map directly from the X-ray diffraction data, read from an mtz, hkl, fcf or mmcif file.
Coot provides extensive features for model building and refinement - i.e. adjusting the model to better fit the electron density, and for validation - i.e. checking that the atomic model agrees with the experimentally derived electron density and makes chemical sense. The most important of these tools is the real space refinement engine, which will optimise the fit of a section of atomic model to the electron density in real time, with graphical feedback. The user may also intervene in this process, dragging the atoms into the right places if the initial model is too far away from the corresponding electron density.
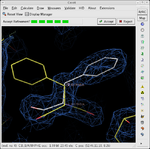
 Tools for general model building:
Tools for general model building:
Tools for moving existing atoms:
Tools for adding atoms to the model:
 In macromolecular crystallography, the observed data is often weak and the observation-to-parameter ratio near 1. As a result it is possible to build an incorrect atomic model into the electron in some cases. To avoid this, careful validation is required. Coot provides a range of validation tools, listed below. Having built an initial model, it is usual to check all of these and reconsider any parts of the model which are highlighted as problematic before deposition of the atomic coordinates with a public database.
In macromolecular crystallography, the observed data is often weak and the observation-to-parameter ratio near 1. As a result it is possible to build an incorrect atomic model into the electron in some cases. To avoid this, careful validation is required. Coot provides a range of validation tools, listed below. Having built an initial model, it is usual to check all of these and reconsider any parts of the model which are highlighted as problematic before deposition of the atomic coordinates with a public database.
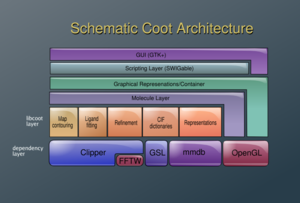 Coot build upon a number of libraries. Crystallographic tools include the Clipper library for manipulating electron density and providing crystallographic algorithms, and the MMDB for the manipulation of atomic models. Other dependencies include FFTW
Coot build upon a number of libraries. Crystallographic tools include the Clipper library for manipulating electron density and providing crystallographic algorithms, and the MMDB for the manipulation of atomic models. Other dependencies include FFTW
, and the GNU Scientific Library
.
Much of the program functionality is available through a scripting interface, which provides access from both the Python and Guile scripting languages.
is a related project with which Coot shares some code. The projects are focussed on slightly different problems, with CCP4mg dealing with presentation graphics and movies, whereas Coot deals with model building and validation.
3D computer graphics
3D computer graphics are graphics that use a three-dimensional representation of geometric data that is stored in the computer for the purposes of performing calculations and rendering 2D images...
. It is primary focussed on the building and validation of atomic models into 3-dimensional electron density
Resolution (electron density)
Resolution in terms of electron density is a measure of the resolvability in the electron density map of a molecule. In X-ray crystallography, resolution is the highest resolvable peak in the diffraction pattern...
maps obtained by X-ray crystallography
X-ray crystallography
X-ray crystallography is a method of determining the arrangement of atoms within a crystal, in which a beam of X-rays strikes a crystal and causes the beam of light to spread into many specific directions. From the angles and intensities of these diffracted beams, a crystallographer can produce a...
methods, although it has also been applied to data from electron microscopy.
Overview
Coot displays electron density maps and atomic models and allows model manipulations such as idealization, real space refinement, manual rotation/translation, rigid-body fitting, ligand search, solvation, mutations, rotamers, and Ramachandran idealizationRamachandran plot
-Introduction and early history:A Ramachandran plot , originally developed in 1963 by G. N. Ramachandran C. Ramakrishnan and V...
. The software is designed to be easy-to-learn for novice users, achieved by ensuring that tools for common tasks are 'discoverable' through familiar user interface elements (menus and toolbars), or by intuitive behaviour (mouse controls). Recent developments have enhanced the usability of the software for expert users, with customisable key bindings, extensions, and an extensive scripting interface.
Coot is free software
Free software
Free software, software libre or libre software is software that can be used, studied, and modified without restriction, and which can be copied and redistributed in modified or unmodified form either without restriction, or with restrictions that only ensure that further recipients can also do...
, distributed under the GNU GPL. It is available from the Coot web site at the University of York. Pre-compiled binaries are available for Linux and Windows from York, and for Mac OS X through Fink
Fink
The Fink project is an effort to port and package open-source Unix programs to Mac OS X. Fink uses dpkg and APT , as well as its own frontend program, fink ....
. Additional support is available through the Coot wiki.
The primary author is Paul Emsley (University of Oxford). Other contributors include Kevin Cowtan, Bernhard Lohkamp and Stuart McNicholas (University of York) and William Scott and Eugene Krissinel.
Features
Coot can be used to read files containing 3D atomic coordinate models of macromolecular structures in a number of formats, including pdbProtein Data Bank (file format)
The Protein Data Bank file format is a textual file format describing the three dimensional structures of molecules held in the Protein Data Bank. The pdb format accordingly provides for description and annotation of protein and nucleic acid structures including atomic coordinates, observed...
, mmcif, and Shelx files. The model may then be rotated in 3D and viewed from any viewpoint. The atomic model is represented by default using a stick-model, with vectors representing chemical bonds. The two halves of each bond are coloured according to the element of the atom at that end of the bond, allowing chemical structure and identity to be visualised in a manner familiar to most chemists.
Coot can also display electron density, which is the result of structure determination experiments such as X-ray crystallography and EM reconstruction. The density is contoured using a 3D-mesh. The contour level controlled using the mouse wheel for easy manipulation - this provides a simple way for the user to get an idea of the 3D electron density profile without the visual clutter of multiple contour levels. Electron density may be read into the program from ccp4
CCP4 (file format)
The CCP4 file format is file generated by the Collaborative Computational Project Number 4 in 1979. The file format for electron density has become industry standard in X-ray crystallography and Cryo-electron microscopy where the result of the technique is a three-dimensional grid of voxels each...
or cns
Crystallography and NMR system
CNS or Crystallography and NMR system, is a software library for computational structural biology. It is an offshoot of X-PLOR and uses much of the same syntax. It is used in the fields of X-ray crystallography and NMR analysis....
map formats, how it is more normal to calculate an electron density map directly from the X-ray diffraction data, read from an mtz, hkl, fcf or mmcif file.
Coot provides extensive features for model building and refinement - i.e. adjusting the model to better fit the electron density, and for validation - i.e. checking that the atomic model agrees with the experimentally derived electron density and makes chemical sense. The most important of these tools is the real space refinement engine, which will optimise the fit of a section of atomic model to the electron density in real time, with graphical feedback. The user may also intervene in this process, dragging the atoms into the right places if the initial model is too far away from the corresponding electron density.
Model building tools
- C-alpha baton mode - trace the main chain of a protein by placing correctly spaced alpha-carbon atoms.
- Ca Zone -> Mainchain - convert an initial trace of the alpha-carbon atoms to a full main-chain trace.
- Place helix here - fit a sequence of amino acids in alpha helixAlpha helixA common motif in the secondary structure of proteins, the alpha helix is a right-handed coiled or spiral conformation, in which every backbone N-H group donates a hydrogen bond to the backbone C=O group of the amino acid four residues earlier...
conformation into density. - Place strand here - fit a sequence of amino acids in beta strand conformation into density.
- Ideal DNA/RNA - build an ideal DNA or RNA fragment.
- Find ligands - find and fit a model to any small molecule which may be bound to the macromolecule.
Tools for moving existing atoms:
- Real space refine zone - optimize the fit of the model to the electron density, while preserving stereochemistry.
- Regularize zone - optimize stereochemistry.
- Rigid body fit zone - optimize the fit of a rigid body to the electron density.
- Rotate/translate zone - manually position a rigid body.
- Rotamer tools (auto fit rotamer, manual rotamer, mutate and autofit, simple mutate)
- Torsion editing (edit chi angles, edit main chain torsions, general torsions)
- Other protein tools (flip peptide, flip sidechain, cis <-> trans)
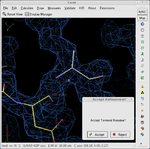
Tools for adding atoms to the model:
- Find waters - add ordered solvent molecules to the model
- Add terminal residue - extend a protein or nucleotide chain
- Add alternate conformation
- Place atom at pointer
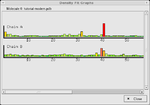
Validation tools
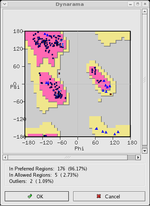
- Ramachandran plotRamachandran plot-Introduction and early history:A Ramachandran plot , originally developed in 1963 by G. N. Ramachandran C. Ramakrishnan and V...
- validate the torsion angles of a protein chain. - Kleywegt plot - examine differences between the torsions of NCS-related chains.
- Incorrect chiral volumes - check for chiral centres with the wrong handedness.
- Unmodelled blobs - check for electron density not accounted for by existing atoms.
- Difference mapDifference density mapIn protein crystallography, a difference density map shows the spatial distribution of the difference between the measured electron density of the crystal and the electron density explained by the current model....
peaks - check for large differences between observed and calculated density. - Check/Delete waters - check for water molecules which do not fit the density.
- Check waters by difference map variance
- Geometry analysis - check for improbable bond lengths, angles, etc.
- PeptidePeptide bondThis article is about the peptide link found within biological molecules, such as proteins. A similar article for synthetic molecules is being created...
omega analysis - check for non-planar peptide bonds. - Temperature factor variance analysis -
- GLN and ASN B-factor outliers -
- Rotamer analysis - check for unusual protein side-chain conformations.
- Density fit analysis - identify parts of the model which don't fit the density.
- Probe clashes - check for Hydrogen atoms with inappropriate environments (using Molprobity).
- NCS differences - check for general differences between NCS related chains.
- Pukka puckers - check for unusual DNA/RNA conformations.
Program architecture
FFTW
FFTW, for "Fastest Fourier Transform in the West", is a software library for computing discrete Fourier transforms , developed by Matteo Frigo and Steven G. Johnson at the Massachusetts Institute of Technology....
, and the GNU Scientific Library
GNU Scientific Library
In computing, the GNU Scientific Library is a software library written in the C programming language for numerical calculations in applied mathematics and science...
.
Much of the program functionality is available through a scripting interface, which provides access from both the Python and Guile scripting languages.
Relation to CCP4mg
The CCP4mg molecular graphics software from Collaborative Computational Project Number 4Collaborative Computational Project Number 4
The Collaborative Computational Project Number 4 in protein crystallography or was set up in 1979 in the United Kingdom to support collaboration between researchers working in software development and assemble a comprehensive collection of software for structural biology...
is a related project with which Coot shares some code. The projects are focussed on slightly different problems, with CCP4mg dealing with presentation graphics and movies, whereas Coot deals with model building and validation.