
Tay-Sachs disease
Overview
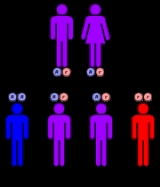
Tay–Sachs disease is an autosomal recessive genetic disorder
. In its most common variant, known as infantile Tay–Sachs disease, it causes a relentless deterioration of mental and physical abilities that commences around six months of age and usually results in death by the age of four.
It is caused by a genetic defect in a single gene with one defective copy of that gene inherited from each parent.
Genetic disorder
A genetic disorder is an illness caused by abnormalities in genes or chromosomes, especially a condition that is present from before birth. Most genetic disorders are quite rare and affect one person in every several thousands or millions....
. In its most common variant, known as infantile Tay–Sachs disease, it causes a relentless deterioration of mental and physical abilities that commences around six months of age and usually results in death by the age of four.
It is caused by a genetic defect in a single gene with one defective copy of that gene inherited from each parent.
Unanswered Questions