
Mitochondrial trifunctional protein deficiency
Encyclopedia
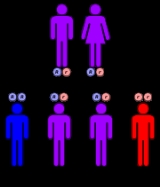
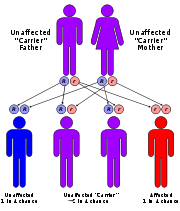
Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats to energy, particularly during periods without food . People with this disorder have inadequate levels of an enzyme that breaks down a certain group of fats called long-chain fatty acids.
 Mutations in the HADHA
Mutations in the HADHA
and HADHB
genes cause mitochondrial trifunctional protein deficiency. These genes each provide instructions for making part of an enzyme complex called mitochondrial trifunctional protein. This enzyme complex functions in mitochondria, the energy-producing centers within cells. As the name suggests, mitochondrial trifunctional protein contains three enzymes that each perform a different function. This enzyme complex is required to break down (metabolize) a group of fats called long-chain fatty acids. Long-chain fatty acids are found in foods such as milk and certain oils. These fatty acids are stored in the body's fat tissues. Fatty acids are a major source of energy for the heart and muscles. During periods of fasting, fatty acids are also an important energy source for the liver and other tissues.
Mutations in the HADHA or HADHB genes that cause mitochondrial trifunctional protein deficiency disrupt all three functions of this enzyme complex. Without enough of this enzyme complex, long-chain fatty acids from food and body fat cannot be metabolized and processed. As a result, these fatty acids are not converted to energy, which can lead to some features of this disorder, such as lethargy and hypoglycemia. Long-chain fatty acids or partially metabolized fatty acids may also build up and damage the liver, heart, and muscles. This abnormal buildup causes the other signs and symptoms of mitochondrial trifunctional protein deficiency.
), muscle weakness (hypotonia
), liver problems, and a high risk for complications such as life-threatening heart
and breathing problems, coma, and sudden unexpected death. The late-onset form is usually less severe; signs and symptoms can include hypotonia, muscle pain, a breakdown of muscle tissue, and abnormalities in the nervous system that affect arms and legs (peripheral neuropathy). Episodes of mitochondrial trifunctional protein deficiency can be triggered by periods of fasting or by illnesses such as viral infections. Diagnosis of this disorder is often confirmed using tandem mass spectrometry
.
Signs and symptoms
Signs and symptoms of mitochondrial trifunctional protein deficiency may begin during infancy or later in life. Features that occur during infancy include feeding difficulties, lack of energy (lethargy), low blood sugar (hypoglycemia), weak muscle tone (hypotonia), and liver problems. Infants with this disorder are also at high risk for serious heart problems, breathing difficulties, coma, and sudden death. Signs and symptoms of mitochondrial trifunctional protein deficiency that may begin after infancy include hypotonia, muscle pain, a breakdown of muscle tissue, and a loss of sensation in the extremities (peripheral neuropathy). Some patients with MTP deficiency show a protracted progressive course associated with myopathy, recurrent rhabdomyolysis, and sensorimotor axonal neuropathy. These patients tend to survive into adolescence and adulthood.Genetics
HADHA
HADHA is a gene associated with long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency.-See also:* Mitochondrial trifunctional protein...
and HADHB
HADHB
Trifunctional enzyme subunit beta, mitochondrial also known as 3-ketoacyl-CoA thiolase, acetyl-CoA acyltransferase, or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene....
genes cause mitochondrial trifunctional protein deficiency. These genes each provide instructions for making part of an enzyme complex called mitochondrial trifunctional protein. This enzyme complex functions in mitochondria, the energy-producing centers within cells. As the name suggests, mitochondrial trifunctional protein contains three enzymes that each perform a different function. This enzyme complex is required to break down (metabolize) a group of fats called long-chain fatty acids. Long-chain fatty acids are found in foods such as milk and certain oils. These fatty acids are stored in the body's fat tissues. Fatty acids are a major source of energy for the heart and muscles. During periods of fasting, fatty acids are also an important energy source for the liver and other tissues.
Mutations in the HADHA or HADHB genes that cause mitochondrial trifunctional protein deficiency disrupt all three functions of this enzyme complex. Without enough of this enzyme complex, long-chain fatty acids from food and body fat cannot be metabolized and processed. As a result, these fatty acids are not converted to energy, which can lead to some features of this disorder, such as lethargy and hypoglycemia. Long-chain fatty acids or partially metabolized fatty acids may also build up and damage the liver, heart, and muscles. This abnormal buildup causes the other signs and symptoms of mitochondrial trifunctional protein deficiency.
Pathophysiology
The mitochondrial trifunctional protein, composed of 4 alpha and 4 beta subunits, catalyzes 3 steps in mitochondrial beta-oxidation of fatty acids: long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), long-chain enoyl-CoA hydratase, and long-chain thiolase activities. Trifunctional protein deficiency is characterized by decreased activity of all 3 enzymes. Clinically, classic trifunctional protein deficiency usually results in sudden unexplained infant death (SIDS; 272120), a Reye-like syndrome, cardiomyopathy, and/or skeletal myopathy.Diagnosis
Onset of this disorder may begin during infancy or later in life. Signs and symptoms of the early onset form can include feeding difficulties, lack of energy (lethargy), low blood sugar (hypoglycemiaHypoglycemia
Hypoglycemia or hypoglycæmia is the medical term for a state produced by a lower than normal level of blood glucose. The term literally means "under-sweet blood"...
), muscle weakness (hypotonia
Hypotonia
Hypotonia is a state of low muscle tone , often involving reduced muscle strength. Hypotonia is not a specific medical disorder, but a potential manifestation of many different diseases and disorders that affect motor nerve control by the brain or muscle strength...
), liver problems, and a high risk for complications such as life-threatening heart
Heart
The heart is a myogenic muscular organ found in all animals with a circulatory system , that is responsible for pumping blood throughout the blood vessels by repeated, rhythmic contractions...
and breathing problems, coma, and sudden unexpected death. The late-onset form is usually less severe; signs and symptoms can include hypotonia, muscle pain, a breakdown of muscle tissue, and abnormalities in the nervous system that affect arms and legs (peripheral neuropathy). Episodes of mitochondrial trifunctional protein deficiency can be triggered by periods of fasting or by illnesses such as viral infections. Diagnosis of this disorder is often confirmed using tandem mass spectrometry
Tandem mass spectrometry
Tandem mass spectrometry, also known as MS/MS or MS2, involves multiple steps of mass spectrometry selection, with some form of fragmentation occurring in between the stages.-Tandem MS instruments:...
.