

Root mean square deviation
Encyclopedia
The root-mean-square deviation (RMSD) is the measure of the average distance between the atoms (usually the backbone atoms) of superimposed proteins. In the study of globular protein conformations, one customarily measures the similarity in three-dimensional structure by the RMSD of the Cα atomic coordinates after optimal rigid body superposition.
When a dynamical system fluctuates about some well-defined average position the RMSD from the average over time can be referred to as the RMSF or root-mean-square fluctuation. The size of this fluctuation can be measured, for example using Mössbauer spectroscopy or nuclear magnetic resonance
, and can provide important physical information. The Lindemann index is a method of placing the RMSF in the context of the parameters of the system.
A widely used way to compare the structures of biomolecules or solid bodies is to translate and rotate one structure with respect to the other to minimize the RMSD. Coutsias, et al. presented a simple derivation, based on quaternion
s, for the optimal solid body transformation (rotation-translation) that minimizes the RMSD between two sets of vectors. They proved that the quaternion method is equivalent to the well-known Kabsch algorithm
.
where δ is the distance between N pairs of equivalent atoms (usually Cα and sometimes C,N,O,Cβ).
Normally a rigid superposition which minimizes the RMSD is performed, and this minimum is returned. Given two sets of points
points  and
and  , the RMSD is defined as follows:
, the RMSD is defined as follows:
When a dynamical system fluctuates about some well-defined average position the RMSD from the average over time can be referred to as the RMSF or root-mean-square fluctuation. The size of this fluctuation can be measured, for example using Mössbauer spectroscopy or nuclear magnetic resonance
Nuclear magnetic resonance
Nuclear magnetic resonance is a physical phenomenon in which magnetic nuclei in a magnetic field absorb and re-emit electromagnetic radiation...
, and can provide important physical information. The Lindemann index is a method of placing the RMSF in the context of the parameters of the system.
A widely used way to compare the structures of biomolecules or solid bodies is to translate and rotate one structure with respect to the other to minimize the RMSD. Coutsias, et al. presented a simple derivation, based on quaternion
Quaternion
In mathematics, the quaternions are a number system that extends the complex numbers. They were first described by Irish mathematician Sir William Rowan Hamilton in 1843 and applied to mechanics in three-dimensional space...
s, for the optimal solid body transformation (rotation-translation) that minimizes the RMSD between two sets of vectors. They proved that the quaternion method is equivalent to the well-known Kabsch algorithm
Kabsch algorithm
The Kabsch algorithm, named after Wolfgang Kabsch, is a method for calculating the optimal rotation matrix that minimizes the RMSD between two paired sets of points...
.
The equation
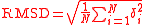
where δ is the distance between N pairs of equivalent atoms (usually Cα and sometimes C,N,O,Cβ).
Normally a rigid superposition which minimizes the RMSD is performed, and this minimum is returned. Given two sets of
points and , the RMSD is defined as follows:-
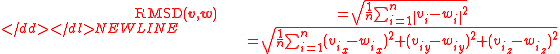
An RMSD value is expressed in length units. The most commonly used unit in structural biologyStructural biologyStructural biology is a branch of molecular biology, biochemistry, and biophysics concerned with the molecular structure of biological macromolecules, especially proteins and nucleic acids, how they acquire the structures they have, and how alterations in their structures affect their function...
is the ÅngströmÅngströmThe angstrom or ångström, is a unit of length equal to 1/10,000,000,000 of a meter . Its symbol is the Swedish letter Å....
(Å) which is equal to 10–10m.
Uses
Typically RMSD is used to make a quantitative comparison between the structure of a partially folded protein and the structure of the native state. For example, the CASPCASPCASP, which stands for Critical Assessment of Techniques for Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994...
protein structure predictionProtein structure predictionProtein structure prediction is the prediction of the three-dimensional structure of a protein from its amino acid sequence — that is, the prediction of its secondary, tertiary, and quaternary structure from its primary structure. Structure prediction is fundamentally different from the inverse...
competition uses RMSD as one of its assessments of how well a submitted structure matches the native state.
Also some scientists who study protein foldingProtein foldingProtein folding is the process by which a protein structure assumes its functional shape or conformation. It is the physical process by which a polypeptide folds into its characteristic and functional three-dimensional structure from random coil....
simulations use RMSD as a reaction coordinateReaction coordinateIn chemistry, a reaction coordinate is an abstract one-dimensional coordinate which represents progress along a reaction pathway. It is usually a geometric parameter that changes during the conversion of one or more molecular entities....
to quantify where the protein is between the folded state and the unfolded state.
See also
- Root mean square deviationRoot mean square deviationThe root-mean-square deviation is the measure of the average distance between the atoms of superimposed proteins...
- Root mean square fluctuation
- QuaternionQuaternionIn mathematics, the quaternions are a number system that extends the complex numbers. They were first described by Irish mathematician Sir William Rowan Hamilton in 1843 and applied to mechanics in three-dimensional space...
– used to optimise RMSD calculations - Kabsch algorithmKabsch algorithmThe Kabsch algorithm, named after Wolfgang Kabsch, is a method for calculating the optimal rotation matrix that minimizes the RMSD between two paired sets of points...
– an algorithm used to minimize the RMSD by first finding the best rotation - GDTGlobal distance testThe global distance test or GDT is a measure of similarity between two protein structures with identical amino acid sequences but different tertiary structures...
– a different structure comparison measure - TM-ScoreTemplate Modeling Score (bioinformatics)The Template Modeling Score or TM-score is a measure of similarity between two protein structures with different tertiary structures. The TM-score is intended as a more accurate measure of the quality of full-length protein structures than the often used RMSD and GDT measures...
– a different structure comparison measure
Further reading
- Shibuya T (2009). "Searching Protein 3-D Structures in Linear Time." Proc. 13th Annual International Conference on Research in Computational Molecular Biology (RECOMB 2009), LNCS 5541:1–15.
External links
- Molecular Distance Measures—a tutorial on how to calculate RMSD
- RMSD—another tutorial on how to calculate RMSD with example code
- Secondary Structure Matching (SSM) — a tool for protein structure comparison. Uses RMSD.
- SuperPose — a protein superposition server. Uses RMSD.
- superpose — structural alignment based on secondary structure matching. By the CCP4 project. Uses RMSD.
- Root mean square deviation