
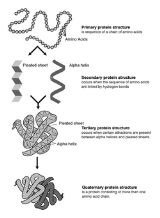
Protein structure
Overview
Organism
In biology, an organism is any contiguous living system . In at least some form, all organisms are capable of response to stimuli, reproduction, growth and development, and maintenance of homoeostasis as a stable whole.An organism may either be unicellular or, as in the case of humans, comprise...
s. Proteins are polymers of amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
s. Classified by their physical size, proteins are nanoparticle
Nanoparticle
In nanotechnology, a particle is defined as a small object that behaves as a whole unit in terms of its transport and properties. Particles are further classified according to size : in terms of diameter, coarse particles cover a range between 10,000 and 2,500 nanometers. Fine particles are sized...
s (definition: 1–100 nm). Each protein polymer – also known as a polypeptide – consists of a sequence formed from 20 possible L-α-amino acids, also referred to as residues. For chains under 40 residues the term peptide
Peptide
Peptides are short polymers of amino acid monomers linked by peptide bonds. They are distinguished from proteins on the basis of size, typically containing less than 50 monomer units. The shortest peptides are dipeptides, consisting of two amino acids joined by a single peptide bond...
is frequently used instead of protein.