
Computational chemistry
Overview
Chemistry
Chemistry is the science of matter, especially its chemical reactions, but also its composition, structure and properties. Chemistry is concerned with atoms and their interactions with other atoms, and particularly with the properties of chemical bonds....
that uses principles of computer science
Computer science
Computer science or computing science is the study of the theoretical foundations of information and computation and of practical techniques for their implementation and application in computer systems...
to assist in solving chemical problems. It uses the results of theoretical chemistry
Theoretical chemistry
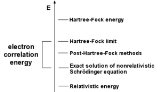
Theoretical chemistry seeks to provide theories that explain chemical observations. Often, it uses mathematical and computational methods that, at times, require advanced knowledge. Quantum chemistry, the application of quantum mechanics to the understanding of valency, is a major component of...
, incorporated into efficient computer program
Computer program
A computer program is a sequence of instructions written to perform a specified task with a computer. A computer requires programs to function, typically executing the program's instructions in a central processor. The program has an executable form that the computer can use directly to execute...
s, to calculate the structures and properties of molecule
Molecule
A molecule is an electrically neutral group of at least two atoms held together by covalent chemical bonds. Molecules are distinguished from ions by their electrical charge...
s and solids. Its necessity arises from the well-known fact that apart from relatively recent results concerning the hydrogen molecular ion (see references therein for more details), the quantum n-body problem
N-body problem
The n-body problem is the problem of predicting the motion of a group of celestial objects that interact with each other gravitationally. Solving this problem has been motivated by the need to understand the motion of the Sun, planets and the visible stars...
cannot be solved analytically, much less in closed form.
Unanswered Questions